
Personalizing Lipid Targets – Beyond PCSK9 Inhibitors
With the release of the 2018 American College of Cardiology/American Heart Association (ACC/AHA) cholesterol guideline, there has been growing enthusiasm in personalized lipid and risk factor management. There are several lipid-lowering agents, including statins, ezetimibe, and recently PCSK9 inhibitors, that have been shown to reduce atherosclerotic cardiovascular disease (ASCVD) risk.1 Statins underwent a major paradigm shift from initial use in reduction of LDL-C for patients with familial hypercholesterolemia (FH), to modulation of ASCVD risk and primary prevention in the general population.
Almost one-third of Americans are on a statin; however, up to 15% of patients report that they are intolerant to the first statin they have been prescribed.2 Moreover, approximately half of patients prescribed statins have been shown to not reach optimal reduction (>40%) in their LDL-C two years after statin initiation.3 Similar challenges controlling LDL-C have been observed in the US and elsewhere. In many patients, ASCVD risk remains persistently high despite lifestyle optimization and treatment with these agents. This has prompted research efforts for new therapies to reduce residual CV risk. Many new drugs for dyslipidemia have been developed from studies in cardiovascular genetics. Here, we summarize a recent article by Hegele and Tsimikas which focuses on novel lipid-lowering agents beyond PCSK9 inhibitors.4
Patients with FH have a high risk of premature CVD. The estimated prevalence of American adults with FH is one in 250.5 There are multiple mechanisms by which FH can be intervened upon. Currently the International Atherosclerosis Society (IAS) guidelines recommend lifestyle and dietary modifications, with high-intesity statin plus ezetimibe as first line. In patients with heterozygous FH, statin monotherapy has been shown to lower the risk of coronary artery disease and mortality by 44%.6 Other studies have corroborated that high dose-statin-ezetimibe combination has resulted in reduction in CV events in these patients.7 Then if LDL-C is not at goal or 50% reduction, niacin, a bile acid sequestrant, or a PCSK-9 inhibitor should be added based on cost and availability according to the IAS. Of note, these recommendations are in conflict with the ACC/AHA guidelines wherein a PCSK9 inhibitor is preferred over niacin or bile acid sequestrants. Up to 80% of FH patients on statin therapy with a PCSK9 inhibitor have been reported as achieving target LDL-C.8,9 If still, LDL-C is not at goal or with <50% reduction, then apheresis, lomitapide or mipomersen may be considered.10
Lomitapide is an inhibitor of microsomal triglyceride transfer protein (MTP), which is responsible for assembling apoB-containing lipoproteins.4 MTP inhibition reduces apoB levels in patients with FH, who lack functional LDL receptors. Mipomersen, an antisense oligonucleotide complementary of apoB mRNA and thereby inhibits apo-B100, is also used in FH.11 Lastly, ATP citrate lyase (ACL) plays a role in cholesterol synthesis. Inhibition of ACL decreases cholesterol synthesis. Bempedoic acid, a newer agent, works through inhibition of ACL and activation of AMP to reduce LDL-C.4 Ongoing gene therapies are also in development. Table 1 and Figure 1 summarize these and other classes of lipid-lowering drugs.
Table 1
Drug |
Mechanism |
Indication |
Stage |
Lipid effect |
Lomitapide |
MTP inhibition |
FH |
Approved |
Reduces LDL-C and TG |
Mipomersen |
apoB inhibition |
FH |
Approved |
Reduces LDL-C |
Bempedoic acid |
ACL inhibition |
Hypercholesterolemia |
Phase 2-3 |
Reduces LDL-C |
Alipogene tiparvovec |
LPL gene therapy |
FCS |
Approved |
Reduces TG |
Volanesorsen |
APOC-3 inhibition |
FCS |
Not approved |
Reduces TG |
Evinacumab |
ANGPTL3 antibody |
Hypercholesterolemia; hypertriglyceridemia |
Phase 2-3 |
Reduces TG, LDL-C, and HDL-C |
IONIS-APO(a) |
Apo(a) |
Anti-apo(a) ASO |
Phase 2-3 |
Reduces Lp(a) |
CSL-112 |
APOA1 |
Low HDL-C |
Phase 3 |
Raises HDL-C |
ACP-501/MEDI6012 |
LCAT |
LCAT deficiency |
Phase 1 |
Raises HDL-C |
Figure 1
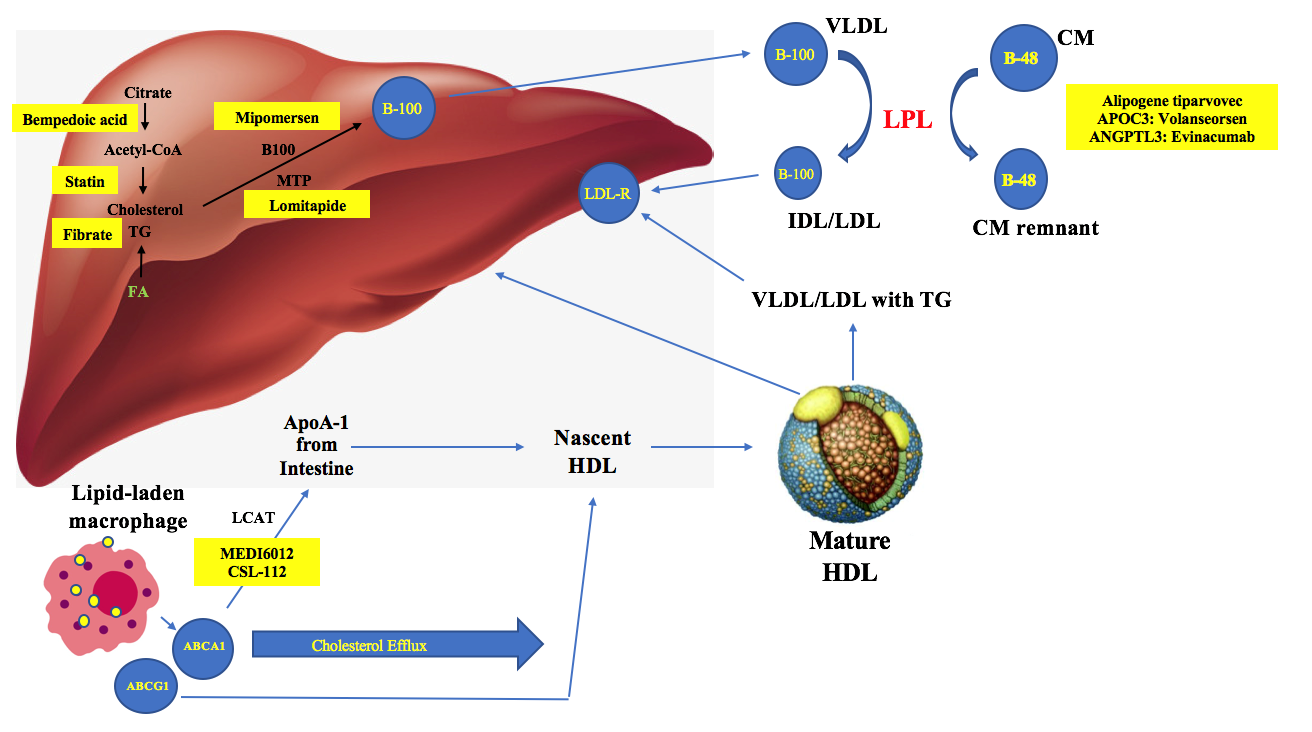
Triglycerides (TG) are also associated with ASCVD risk. However, it has been suggested that TGs themselves do not directly contribute to plaque formation – instead, it is the VLDL particles, remnants and IDLs that play the strongest role. Together, TGs and remnant particles interact to increase atherogenesis. Hypertriglyceridemia may be monogenic (familial chlyomicronemia syndrome, FCS) or from secondary factors. ASCVD risk seems to be higher in patients with secondary hypertriglyceridemia but not with a pure monogenic chylomicronemia.12 While statins are used in in treatment of hypertriglyceridemia, niacin, fibrates and omega-3s have been recommended for patients with hypertriglyceridemia.
Recently, REDUCE-IT (Reduction of Cardiovascular Events With Icosapent Ethyl-Intervention Trial) showed Vascepa (high dose icosapent ethyl, omega-3s) reduced triglycerides, CV events and deaths in patients with known CVD or at risk for developing it, and those already on statins with well-controlled LDL-C.13 The primary composite endopoint in patients taking Vascepa was 17.2% versus 22 percent in patients taking placebo, with an absolute risk reduction of 4.8%. There have been several novel agents trialed for treatment of hypertriglyceridemia, including apolipoprotein C-III (apo C-III) inhibitors and lipoprotein lipase (LPL) gene therapy (Figure 1). Apo C-III is found on chylomicrons and VLDL, and interferes with triglyceride clearance by inhibiting LPL activity.4
Volanesorsen is an antisense oligonucleotide that inhibits translation of Apo-CIII mRNA, thereby lowering triglycerides. It fell short of final FDA approval for FCS and refractory hypertriglyceridemia, due to a small increased risk of thrombocytopenia. Additional targets for hypertriglyceridemia include apoCII, apoA-V and ANGPTL4.4
While HDL-C is an inverse predictor of CV event risk, human genetic studies have failed to show a similar causal relationship between HDL-C and ASCVD risk as they have for LDL-C. In other words, there is no evidence in humans that raising HDL-C results in reduced risk of having an ASCVD event. Cholesteryl ester transfer protein (CETP) transfers cholesterol esters from non-atherogenic HDL to potentially pro-atherogenic non-HDL fraction.
While early studies showed CETP deficiency associated with higher HDL-C and protection from ASCVD, later studies showed equivocal findings.14 Three different CETP inhibitors all failed to reduce ASCVD events in large RCTs. This has led to questions regarding the nature of the HDL hypothesis. One possibility is that HDL function, which can be measured through cholesterol efflux, over quantity, may be more relevant for ASCVD risk assessment.4,14
ApoA-I is another protein related to HDL function. It is found on HDL and chylomicrons. Based on animal models, it has been hypothesized that ApoA-I infusions may work to stabilize plaque and modulate atherosclerosis regression. Over the past few years, several HDL mimetics have made it to clinical trials. From preliminary results, treatment with these agents has failed to produce a significant effect on primary efficacy endpoints. These infusions, however, may be more pertinent for patients with familial hypoalphalipoproteinemia (FHA) and homozygous familial hypercholesterolemia, with small studies underway.
Most novel lipid therapeutics have sprung from studies of rare diseases with severe phenotypes. Cardiovascular outcome research focusing on Mendelian randomization may potently validate therapeutic targets. While new biologics and targeted therapies may represent future second line combination options for select patients, the current most effective action is to maximize preventative measures with lifestyle and evidence based medications.
References
- Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2019;73:e285-350.
- Salami JA, Warraich H, Valero-Elizondo J, et al. National trends in statin use and expenditures in the US adult population from 2002 to 2013: insights from the Medical Expenditure Panel Survey. JAMA Cardiol 2017;2:56-65.
- Akyea RK, Kai J, Qureshi N, Iyen B, Weng SF. Sub-optimal cholesterol response to initiation of statins and future risk of cardiovascular disease. Heart 2019;105:975-81.
- Hegele RA, Tsimikas S. Lipid-lowering agents. Circ Res 2019;124:386-404.
- De Ferranti SD, Rodday Am, Mendelson MM, Wong JB, Leslie LK, Sheldrick RC. Prevalence of familial hypercholesterolemia in the 1999 to 2012 United States National Health and Nutrition Examination Surveys (NHANES). Circulation 2016;133:1067-72.
- Besseling J, Hovingh GK, Huijgen R, Kastelein JJP, Hutten BA. Statins in familial hypercholesterolemia: consequences for coronary artery disease and all-cause mortality. J Am Coll Cardiol 68:252-60.
- Lind S, Olsson AG, Eriksson M, Rudling M, Eggertsen G, Angelin B. Autosomal recessive hypercholesterolaemia: normalization of plasma LDL cholesterol by ezetimibe in combination with statin treatment. J Intern Med 2004;256:406-12.
- Defesche JC, Stefanutti C, Langslet G, et al. Efficacy of alirocumab in 1191 patients with a wide spectrum of mutations in genes causative for familial hypercholesterolemia. J Clin Lipidol 2017;11:1338-46.
- Rosenson RS, Hegele RA, Koenig W. Cholesterol-lowering agents. Circ Res 2019;124:364-85.
- Santos RD, Gidding SS, Hegele RA, et al. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol 2016;4:850-61.
- Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholestlerolaemia: a randomised, double-blind, placebo-controlled trial. Lancet 2010;375:998-1006.
- Dron JS, Wang J, Cao H, et al. Severe hypertriglyceridemia is primarily polygenic. J Clin Lipidol 2019;13:80-8.
- Bhatt DL, Steg PG, Miller M, et al. Effects of icosapent ethyl on total ischemic events: from REDUCE-IT. J Am Coll Cardiol 2019;73:2791-802.
- Hovingh GK, Rader DJ, Hegele RA. HDL re-examined. Curr Opin Lipidol 2015;26:127-32.
Keywords: Apolipoprotein C-III, Cholesterol Ester Transfer Proteins, Hyperlipoproteinemia Type II, Cholesterol Esters, Apolipoprotein A-I, Niacin, Apolipoproteins B, Hydroxymethylglutaryl-CoA Reductase Inhibitors, Lipoprotein Lipase, Hypercholesterolemia, Receptors, LDL, Coronary Artery Disease, Fibric Acids, Apolipoproteins C, Triglycerides, Risk Factors, Chylomicrons, Biological Products, RNA, Messenger, Random Allocation, Lipid Metabolism, Inborn Errors, Oligonucleotides, Eicosapentaenoic Acid, Antibodies, Monoclonal, Benzimidazoles, Atherosclerosis, Hypertriglyceridemia, Blood Component Removal, Oligonucleotides, Antisense, Risk Assessment, Primary Prevention, Life Style, Thrombocytopenia, Genetic Therapy, Bile Acids and Salts, Phenotype, Adenosine Monophosphate, Dyslipidemias
< Back to Listings