
Anatomy, Function, and Dysfunction of the RV
Introduction
New impetus in understanding the right ventricle (RV) in health and disease has emerged from accumulating evidence of its symptomatic and prognostic relevance in multiple clinical scenarios.1 When the clinician faces an abnormal RV, the main underlying processes include pressure overload, volume overload, or a primary myocardial process (Table 1 and Figure 1). The clinical course and therapeutic approach in these three situations differ significantly, although they frequently coexist.2
Table 1: Etiologies of RV Pressure Overload, Volume Overload, and RV Cardiomyopathy
| RV Pressure Overload | RV Volume Overload | RV Cardiomyopathy |
| Pulmonary hypertension (PH)* | Valvular regurgitation | Myocardial infarction (MI) |
| Pulmonary arterial hypertension | Tricuspid | Arrhythmogenic RV cardiomyopathy |
| Due to left heart disease | Pulmonary | Dilated cardiomyopathy |
| Due to lung disease and/or hypoxia | Systemic-to-pulmonary shunt | Hypertrophic cardiomyopathy |
| Chronic thromboembolic PH and other pulmonary artery (PA) obstructions | Atrial septal defect | Amyloidosis |
| Unclear and/or multifactorial mechanisms | Partial anomalous pulmonary vein drainage | Myocarditis |
| Pulmonary valve stenosis | High output states** (i.e., thyrotoxicosis) | Sarcoid |
| PA stenosis | Transplant | |
| Pulmonary embolism | Post-surgery | |
| Post left ventricular (LV) assist device | ||
| Cardiotoxicity (i.e., chemotherapy) | ||
| Sepsis |
Figure 1
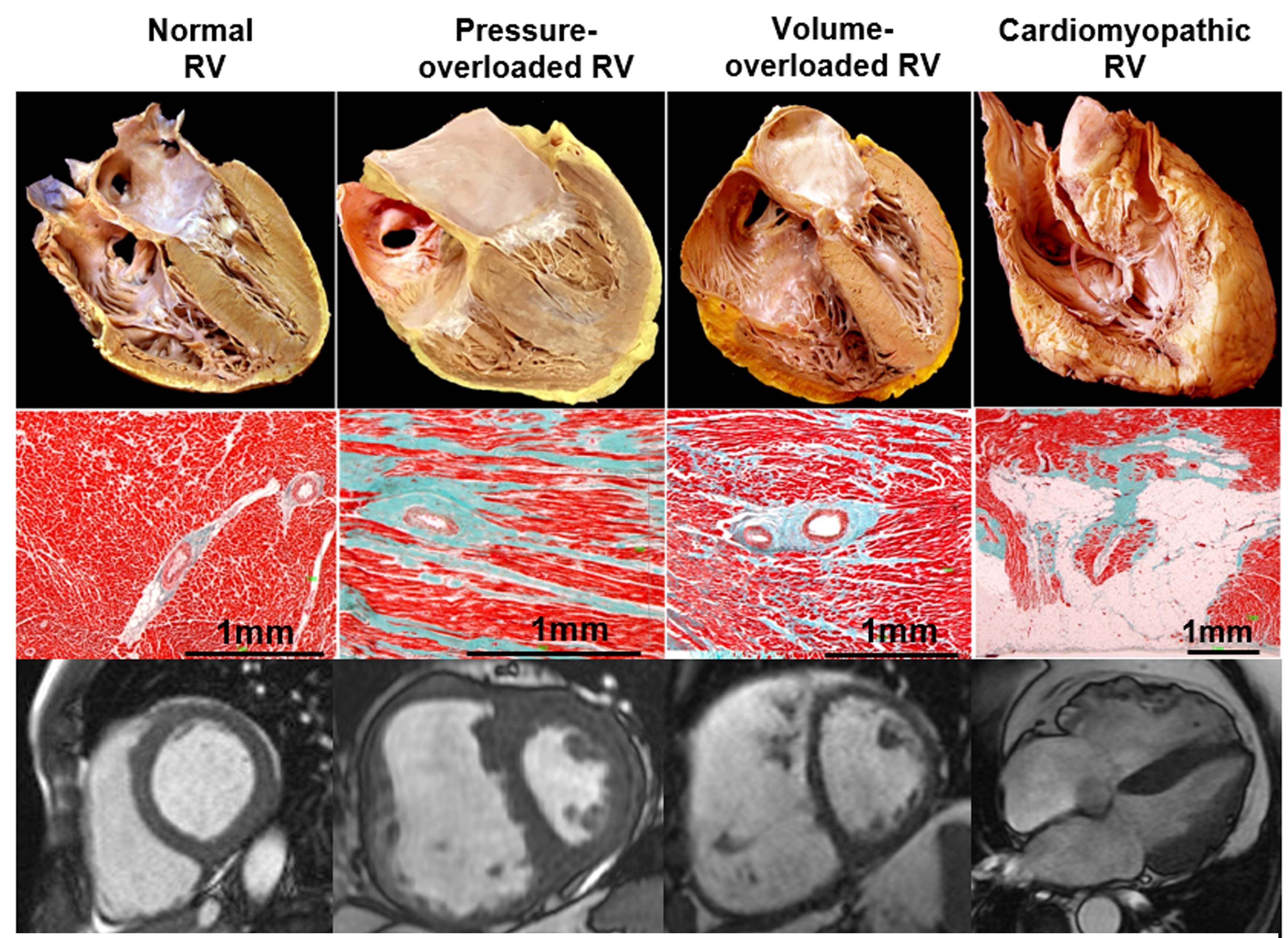
The Normal RV
The RV is 10-15% larger in volume than the LV, with thinner free wall and smaller mass.3 With aging, there is a reduction in RV mass and volume and an increase in ejection fraction (EF). RV volumes and mass tend to be higher in men, whereas RV EF is lower.4 Three anatomic components are often described: 1) the inlet, containing the tricuspid valve apparatus; 2) the trabeculated apex, and 3) the outlet or infundibulum. Although in the LV there are 3 distinct myocardial layers of aggregated cardiomyocytes, there are only 2 in the RV: a superficial, predominantly circumferential layer and a subendocardial, predominantly longitudinal layer (Figure 2). The RV and LV are closely interrelated, not only through the septum, but also through shared epicardial circumferential myocytes and the pericardial space, all of which constitute the anatomic basis for biventricular interdependence.5
Figure 2
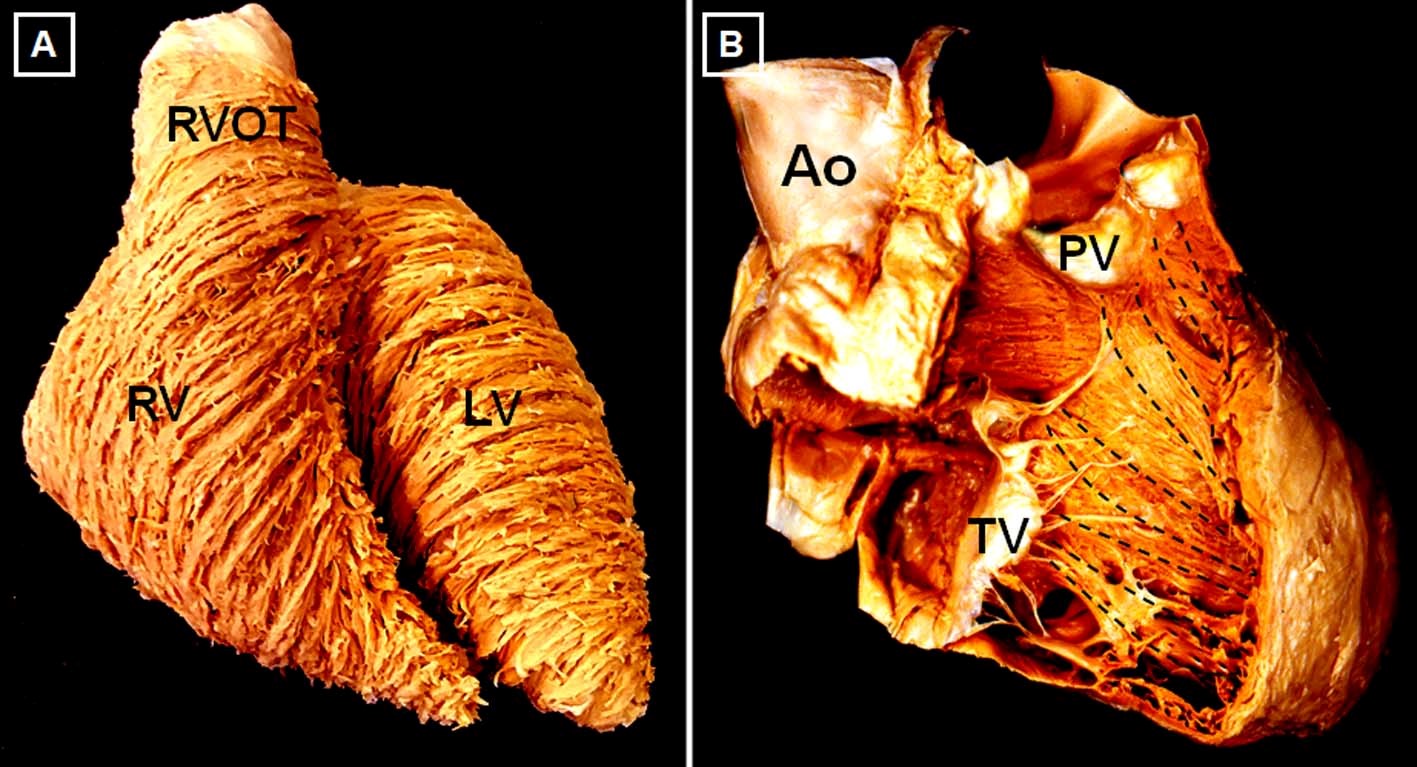
There are no convincing data to suggest that the processes of excitation and contraction or the energy source (predominantly fatty acids) are different in the RV than the LV.6 Resting coronary blood flow is lower in the RV and occurs both in systole and diastole. Oxygen consumption and extraction at rest are also lower, resulting in higher oxygen extraction reserve if needed. Together with prominent collaterals from the left coronary system, these features make the RV relatively resistant to ischemia.7,8 However, because of thinner wall and higher dependence on coronary perfusion pressure, RV perfusion is more vulnerable to increases in RV cavitary (and thus intramural) pressure and to systemic hypotension.9
The RV contracts in a peristalsis-like motion that occurs earlier in the sinus and apex than the conus,3 and longitudinal shortening accounts for approximately 75% of RV contraction.10 The LV is an important contributor to RV ejection, generating 20-40% of RV stroke volume (SV), predominantly but not only via septal contraction.1,6 The thinner wall and lower volume-to-wall surface area ratio render the RV more compliant and capable of accommodating increased preload but unable to cope with brisk increments in PA pressures.1 An acute increase of either preload or afterload is immediately associated with RV dilatation to preserve SV. After several minutes, this heterometric adaptation is replaced by a homeometric adaptation with volume normalization and increased contractility. From invasive pressure loops pulmonary arterial elastance (index of RV afterload), and RV end-systolic elastance (index of RV contractility) can be obtained.5 The comparison of arterial elastance and end-systolic elastance allows determination of the adequacy of adaptation of RV contractility to afterload or "coupling" to the pulmonary circulation. Optimal ventriculoarterial coupling takes place when both elastances are equal (end-systolic elastance / arterial elastance = 1). However, the optimal end-systolic elastance / arterial elastance for ejection at minimal energy cost, as seen in the normal RV, is 1.5-2.6 Strictly, end-systolic elastance / arterial elastance is measured invasively, although simplified invasive and imaging-based noninvasive approaches have been developed.2
The Pressure-Overloaded RV
RV pressure overload, most commonly secondary to PH, leads to RV hypertrophy, predominantly end-systolic and early-diastolic flattening of the interventricular septum, and eventually progressive RV dilation and dysfunction (Figure 1). An oxygen supply-demand imbalance has been demonstrated in PH-associated heart failure, with potential mechanisms including capillary rarefaction, systemic hypotension, impairment of systolic coronary flow due to increased wall stress, mechanical inefficiency, and compression of the right coronary artery by a dilated PA.2 A potential consequence of impaired oxygen supply is metabolic remodeling with "metabolic switch" from fatty acid oxidation to less-efficient but oxygen-sparing anaerobic glycolysis as preferred energy source.11 Acute pressure overload (i.e., pulmonary embolism) leads to acute dilatation with increased wall stress, decreased perfusion, and also metabolic switch. However, as opposed to chronic PH, it is additionally associated with inflammatory leucocyte influx of the myocardium, and myocyte lysis particularly at the outflow tract.12
In chronic pressure overload, the RV initially responds with adaptive remodeling: relatively preserved volumes and function with compensatory concentric hypertrophy. When contractility can no longer increase to match afterload, maladaptive remodeling takes place with eccentric hypertrophy, progressive RV dilatation and dyssynchrony, and maintenance of SV through Frank-Starling mechanisms (heterometric adaptation). This comes at the price of increased filling pressures and, eventually, clinical decompensation.6 RV dilatation causes additional deleterious interventricular interactions in diastole (see below).
The Volume-Overloaded RV
The hallmarks are RV dilatation and hypertrophy (increased free wall mass albeit preserved thickness) and predominantly diastolic leftward septal shift (Figure 1). Because of the anatomic and physiological features discussed above, volume overload is much better tolerated than pressure overload.1 RV dilatation leads to augmented wall tension and therefore to simultaneous increase in afterload.
RV contractility remains preserved for a long time, although contractile reserve may be compromised. Volume overload leads to simultaneous LV dysfunction,5 primarily due to underfilling secondary to septal displacement and changes in LV geometry rather than decreased RV-forward SV.1 Chronic volume overload may eventually lead to RV systolic dysfunction and increased morbi-mortality, particularly in the presence of superimposed pressure overload and/or marked RV enlargement, which argues for corrective interventions before significant RV dilatation ensues.1
The Cardiomyopathic RV
Myocardial insults of various etiologies may involve the RV. Its adaptive response in this context differs not only according to the underlying pathologic process, but also, importantly, to any associated volume and/or pressure overload. Ischemia-related RV dysfunction is reviewed below as an example.
Ischemic Heart Disease
Acute RV MI typically occurs when there is occlusion of the right coronary artery proximal to marginal branches.7 Acute RV ischemia and/or MI lead to both systolic and diastolic dysfunction. Decreased RV EF may cause reduced output and hypotension, which in turn exacerbate ischemia. Reduced RV compliance is accentuated by pericardial constraint on a dilated RV, leading to marked increases in right atrial pressures and impaired interventricular interactions. In this situation, adequate RV filling (and SV maintenance) may be highly dependent on both adequate preload, inotropic support, and/or preserved atrial contraction.7,8 RV involvement in acute MI is detected in autopsy or magnetic resonance studies more commonly than clinically (up to 30% and 50%, respectively), particularly in inferior but also often in anterior infarcts.13,14
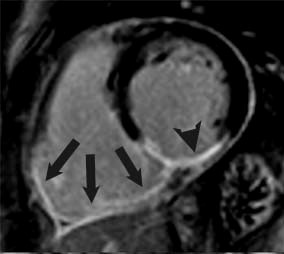
Because of the aforementioned relative resistance to ischemia of the RV, chronic RV infarcts are somewhat infrequent and can be identified in vivo in 5-13% of patients with magnetic resonance (Figure 3).14,15 For the same reasons, RV function tends to recover after the acute phase. Persistent post-infarction RV dysfunction is thus likely multifactorial in etiology, including not only chronic MI but also LV-RV interactions, increased afterload, ischemia, and mitral regurgitation.15
Figure 3
References
- Konstam MA, Kiernan MS, Bernstein D, et al. Evaluation and Management of Right-Sided Heart Failure: A Scientific Statement From the American Heart Association. Circulation 2018;137:e578-e622.
- Sanz J, Sánchez-Quintana D, Bossone E, Bogaard HJ, Naeije R. Anatomy, Function, and Dysfunction of the Right Ventricle: JACC State-of-the-Art Review. J Am Coll Cardiol 2019;73:1463-82.
- Dell'Italia LJ. The right ventricle: anatomy, physiology, and clinical importance. Curr Probl Cardiol 1991;16:653-720.
- Kawut SM, Lima JA, Barr RG, et al. Sex and race differences in right ventricular structure and function: the multi-ethnic study of atherosclerosis-right ventricle study. Circulation 2011;123:2542-51.
- Naeije R, Badagliacca R. The overloaded right heart and ventricular interdependence. Cardiovasc Res 2017;113:1474-85.
- Lahm T, Douglas IS, Archer SL, et al. Assessment of Right Ventricular Function in the Research Setting: Knowledge Gaps and Pathways Forward. An Official American Thoracic Society Research Statement. Am J Respir Crit Care Med 2018;198:e15-e43.
- O'Rourke RA, Dell'Italia LJ. Diagnosis and management of right ventricular myocardial infarction. Curr Probl Cardiol 2004;29:6-47.
- Harjola VP, Mebazaa A, Čelutkienė J, et al. Contemporary management of acute right ventricular failure: a statement from the Heart Failure Association and the Working Group on Pulmonary Circulation and Right Ventricular Function of the European Society of Cardiology. Eur J Heart Fail 2016;18:226-41.
- Zong P, Tune JD, Downey HF. Mechanisms of oxygen demand/supply balance in the right ventricle. Exp Biol Med (Maywood) 2005;230:507-19.
- Brown SB, Raina A, Katz D, Szerlip M, Wiegers SE, Forfia PR. Longitudinal shortening accounts for the majority of right ventricular contraction and improves after pulmonary vasodilator therapy in normal subjects and patients with pulmonary arterial hypertension. Chest 2011;140:27-33.
- Rich S, Pogoriler J, Husain AN, Toth PT, Gomberg-Maitland M, Archer SL. Long-term effects of epoprostenol on the pulmonary vasculature in idiopathic pulmonary arterial hypertension. Chest 2010;138:1234-9.
- Watts JA, Marchick MR, Kline JA. Right ventricular heart failure from pulmonary embolism: key distinctions from chronic pulmonary hypertension. J Card Fail 2010;16:250-9.
- Andersen HR, Falk E, Nielsen D. Right ventricular infarction: frequency, size and topography in coronary heart disease: a prospective study comprising 107 consecutive autopsies from a coronary care unit. J Am Coll Cardiol 1987;10:1223-32.
- Masci PG, Francone M, Desmet W, et al. Right ventricular ischemic injury in patients with acute ST-segment elevation myocardial infarction: characterization with cardiovascular magnetic resonance. Circulation 2010;122:1405-12.
- Sabe MA, Sabe SA, Kusunose K, Flamm SD, Griffin BP, Kwon DH. Predictors and Prognostic Significance of Right Ventricular Ejection Fraction in Patients With Ischemic Cardiomyopathy. Circulation 2016;134:656-65.
Clinical Topics: Acute Coronary Syndromes, Arrhythmias and Clinical EP, Diabetes and Cardiometabolic Disease, Dyslipidemia, Heart Failure and Cardiomyopathies, Prevention, Pulmonary Hypertension and Venous Thromboembolism, Valvular Heart Disease, Atrial Fibrillation/Supraventricular Arrhythmias, Lipid Metabolism, Acute Heart Failure, Pulmonary Hypertension, Hypertension, Mitral Regurgitation
Keywords: Acute Coronary Syndrome, Stroke Volume, Systole, Diastole, Myocytes, Cardiac, Dilatation, Heart Ventricles, Tricuspid Valve, Pulmonary Circulation, Mitral Valve Insufficiency, Coronary Vessels, Glycolysis, Peristalsis, Fatty Acids, Prognosis, Anaerobiosis, Atrial Fibrillation, Atrial Pressure, Myocardium, Heart Failure, Pericardium, Cardiomyopathies, Cardiomyopathies, Magnetic Resonance Spectroscopy, Myocardial Ischemia, Aneurysm, Hypertrophy, Pituitary Gland, Infarction, Hypotension, Fibrosis, Pulmonary Embolism, Oxygen Consumption, Hypertension, Pulmonary
< Back to Listings