
CV Toxicity and Mitochondrial Dysfunction: Why Younger Hearts Are at Risk
Drug-induced cardiovascular toxicity is thought to be mediated by mitochondrial dysfunction.1 Mitophagy is a process of recovery through which damaged mitochondria are removed.2 Recurrent mitochondrial toxicity secondary to drug and radiation therapy can overcome mitophagy and lead to sustained mitochondrial dysfunction.3 In this review, we focus on how various cancer therapies can lead to cardiovascular toxicity by simultaneously regulating mitochondrial DNA (mtDNA) damage, altering mitochondrial reactive oxygen species (mtROS) production, and impairing nuclear DNA damage responses (DDR).
Premature Aging and Sustained mtROS Production in Cancer Survivors
In the St. Jude Lifetime study cohort, survivors of childhood cancer had a high prevalence of chronic health conditions of the lungs, heart, and brain. Interestingly, both cardiotoxic treatment exposed and unexposed survivors showed higher abnormalities in cardiac function, traditional risk factors for atherosclerotic disease, and systemic inflammation compared with the non-cancer group.4 Lipshultz et al. suggested that premature aging induced by cancer treatment is a major contributor to the chronic health problems in cancer survivors.4 However, the molecular mechanisms underlying this premature aging process remain unclear. A common feature of chemoradiation therapy and aging is the shortening of telomeres.5 Telomere shortening has been associated with cardiovascular disease,6 and accounts at least partially for the long-term effects of chemoradiation in young cancer survivors.7 The telomeric DNA sequences TTAGGG are sevenfold more likely to be damaged by H2O2 due to the propensity of iron to bind to these sequences and generate OH radicals via Fe2+-mediated Fenton reactions.8,9 Very recently, Qian et al. have reported the critical role of mtROS in telomere DNA damage but not genomic DNA damage9 and showed that a feedback loop between mitochondria dysfunction and nuclear DDR sustains mtROS production, leading to premature aging via telomere shortening and erosion (Figure 1).
Figure 1
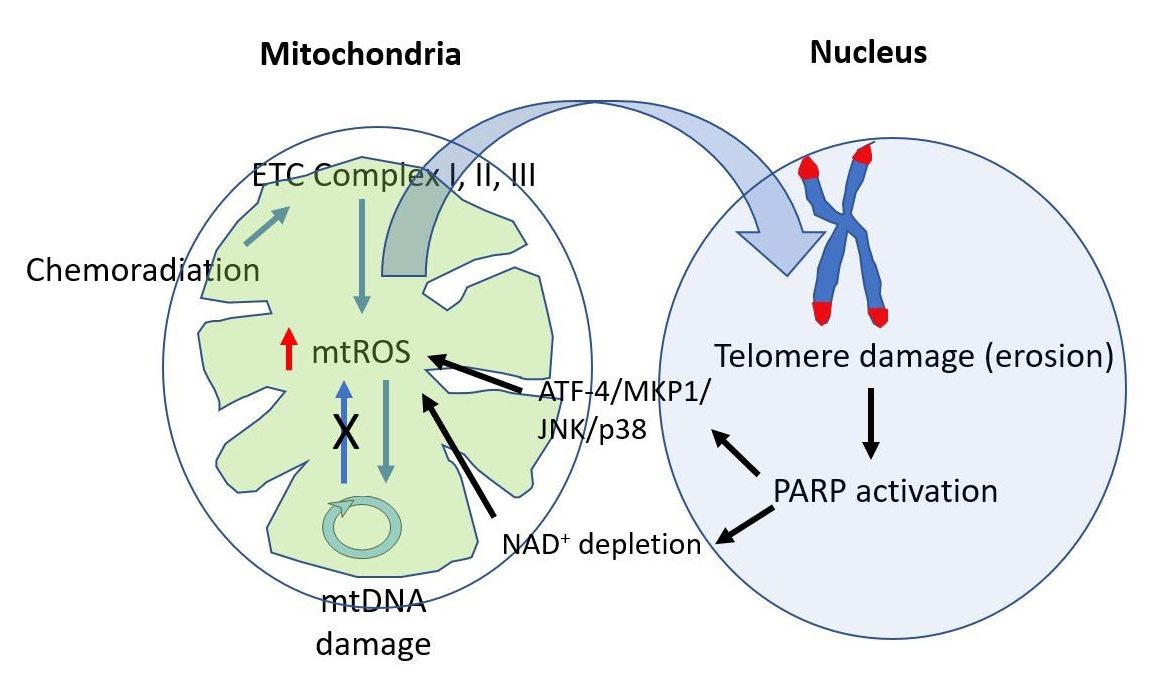
Cardiotoxicity Through Sustained Mitochondrial Dysfunction
Mitochondria are the main sources of cellular energy via the process of respiration and oxidative phosphorylation for generation of adenosine triphosphate (ATP). Oxidative phosphorylation results from two components: the electron transport chain (ETC) generation of proton motive force and chemiosmosis-driven generation of ATP by complex V. The ETC is composed of a series of membrane-embedded proteins that include four large complexes (I-IV) that except for complex II, which is solely nuclear encoded, requires both nuclear DNA and mtDNA. Two key redox cofactors, NADH and FADH2, deliver electrons from tricarboxylic acid cycle to ETC complex I and II, respectively. The electrons pass sequentially through complexes releasing energy to generate a proton motive force via complexes I, III, and IV by pumping protons (H+) across the inner mitochondrial membrane. This electrochemical gradient drives chemiosmosis through complex V (ATP synthase) for generation of ATP. ATP production is critical for cellular energy transfer and regulation of enzymes required for the steps of glycolysis and beta-oxidation.10
The mitochondrial matrix has an approximate pH of 7.8 with anionic charge and unique chemical characteristics that favor the accumulation of xenobiotics in the matrix and in inner mitochondrial membrane.11-13 Scatena et al. have reported that lipophilic compounds of cationic character and weak acids in anionic forms can concentrate in mitochondria. The build-up of these lipophilic and acidic xenobiotics inside the mitochondria can alter or inhibit various mitochondrial function.14-16
Cardiovascular toxic drugs, such as doxorubicin and paclitaxel,17 and radiation18 can alter mitochondrial function in the following ways:
- Inhibition of ETC protein complexes and ATP synthesis
- Inhibition of tricarboxylic acid cycle flux
- Inhibition of various mitochondrial transporters
- Inhibition of mitochondrial transcriptional and translational machinery
- Uncoupling of the ETC from ATP synthesis19
Reactive oxygen species (ROS) generated within the mitochondria cause oxidative mitochondria damage (Figure 1).20-22 Individual mitochondria contain their own DNA (mtDNA), which is a circular intron-free genome. mtDNA is potentially more susceptible to oxidative damage than nuclear DNA because of the following:
- The tendency of lipophilic and charged chemicals to accumulate in the mitochondria
- The proximity of mtDNA to ETC-induced mtROS
- The lack of chromatin packing system including histone, telomere, and minimum DNA repair system compared with nuclear DNA23
Therefore, mtROS can cause significant damage to mtDNA and increase DNA mutations at a rate that is estimated to be at least 10-20 times higher for mtDNA than for nuclear DNA.24
Feedback Loop Between mtDNA Damage and mtROS Theory Is Not Supported by the Study Using mtDNA Mutator Mice
Complexes I, II, and III are the major sites of mtROS, and mtDNA encodes sub-units in two of these three complexes.25,26 It is thus reasonable to assume that the induction of mtDNA mutations induces mtROS by decreasing the activity of complexes I and III, forming a feedback loop sustaining mitochondrial dysfunction.23 However, this theory is not supported experimentally in mice (Figure 1). mtDNA polymerase Γ is the polymerase that is responsible for replication and repair of mtDNA. Therefore, the lack of mtDNA polymerase Γ accumulates mtDNA mutations and deletions and reduces respiratory chain enzyme activities and mitochondrial ATP production in the hearts.27 However, in mtDNA mutator mice, markers of ROS damage were not higher compared with control mice.28,29 Therefore, mtDNA mutations are important to reduce ATP level but may not be crucial for mtROS production.23
The Cross Talk Between mtROS Production and Nuclear DNA Damage Response May Be Crucial for Sustained mtROS Production After Chemoradiation
The key role of ROS in regulating genotoxic stress and nuclear DNA damage caused by chemotherapeutic agents and ionizing radiation is well-established.30 ROS-induced nuclear DNA damage can initiate mtROS production through several potential mechanisms. PARP1 is a highly conserved nuclear enzyme that acts as a zinc-dependent DNA damage sensor that can bind to both single- and double-stranded DNA breaks.31 Hocsak et al. have reported that ROS-induced nuclear DNA damage increases mtROS production via PARP1 activation and subsequent JNK/p38 activation (Figure 1).32 Alternatively, ROS-induced nuclear DNA damage activates PARP or SIRT1, which use NAD+ as a co-factor and deplete NAD+ at the cellular level. The depletion of NAD+ plays a critical role in mitochondria metabolism and decreased mitophagy, leading to more mtROS production by accumulating damaged mitochondria (Figure 1).2 However, the exact molecular mechanism of this NAD+ depletion-mediated mtROS production remains unclear.
Conclusion
In this brief review, we focus on the critical role of mitochondria-nuclear cross talk in regulating sustained mtROS production, which leads to premature aging in cancer survivors. Although the importance of long-term effects of chemoradiation is now evident, this topic is overshadowed by acute effects of chemoradiation in cardiovascular disease. This accelerated premature aging is important not only in young cancer survivors, but also in older patients or patients with pre-existing cardiovascular disease who show worse outcomes after chemoradiation. Therefore, an urgent need exists to determine the molecular mechanisms of premature aging in cancer survivors.
References
- Scatena R. Mitochondria and drugs. Adv Exp Med Biol 2012;942:329-46.
- Fang EF, Scheibye-Knudsen M, Brace LE, et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014;157:882-96.
- Weinhouse C. Mitochondrial-epigenetic crosstalk in environmental toxicology. Toxicology 2017;391:5-17.
- Lipshultz SE, Landy DC, Lopez-Mitnik G, et al. Cardiovascular status of childhood cancer survivors exposed and unexposed to cardiotoxic therapy. J Clin Oncol 2012;30:1050-7.
- Chang L, Weiner LS, Hartman SJ, et al. Breast cancer treatment and its effects on aging. J Geriatr Oncol 2019;10:346-55.
- De Meyer T, Nawrot T, Bekaert S, De Buyzere ML, Rietzschel ER, Andrés V. Telomere Length as Cardiovascular Aging Biomarker: JACC Review Topic of the Week. J Am Coll Cardiol 2018;72:805-13.
- Abe J, Martin JF, Yeh ET. The Future of Onco-Cardiology: We Are Not Just "Side Effect Hunters". Circ Res 2016;119:896-9.
- Ren JG, Xia HL, Just T, Dai YR. Hydroxyl radical-induced apoptosis in human tumor cells is associated with telomere shortening but not telomerase inhibition and caspase activation. FEBS Lett 2001;488:123-32.
- Qian W, Kumar N, Roginskaya V, et al. Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction. Proc Natl Acad Sci U S A 2019;116:18435-44.
- Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res 2013;112:1171-88.
- Murphy MP, Smith RA. Drug delivery to mitochondria: the key to mitochondrial medicine. Adv Drug Deliv Rev 2000;41:235-50.
- Szewczyk A, Wojtczak L. Mitochondria as a pharmacological target. Pharmacol Rev 2002;54:101-27.
- Suski J, Lebiedzinska M, Machado NG, et al. Mitochondrial tolerance to drugs and toxic agents in ageing and disease. Curr Drug Targets 2011;12:827-49.
- Scatena R, Bottoni P, Vincenzoni F, et al. Bezafibrate induces a mitochondrial derangement in human cell lines: a PPAR-independent mechanism for a peroxisome proliferator. Chem Res Toxicol 2003;16:1440-7.
- Scatena R, Bottoni P, Martorana GE, et al. Mitochondrial respiratory chain dysfunction, a non-receptor-mediated effect of synthetic PPAR-ligands: biochemical and pharmacological implications. Biochem Biophys Res Commun 2004;319:967-73.
- Scatena R, Messana I, Martorana GE, et al. Mitochondrial damage and metabolic compensatory mechanisms induced by hyperoxia in the U-937 cell line. J Biochem Mol Biol 2004;37:454-9.
- Guigni BA, Callahan DM, Tourville TW, et al. Skeletal muscle atrophy and dysfunction in breast cancer patients: role for chemotherapy-derived oxidant stress. Am J Physiol Cell Physiol 2018;315:C744-C756.
- Baselet B, Sonveaux P, Baatout S, Aerts A. Pathological effects of ionizing radiation: endothelial activation and dysfunction. Cell Mol Life Sci 2019;76:699-728.
- Will Y, Shields JE, Wallace KB. Drug-Induced Mitochondrial Toxicity in the Geriatric Population: Challenges and Future Directions. Biology (Basel) 2019;8:E32.
- Wei YH, Lu CY, Lee HC, Pang CY, Ma YS. Oxidative damage and mutation to mitochondrial DNA and age-dependent decline of mitochondrial respiratory function. Ann N Y Acad Sci 1998;854:155-70.
- Duchen MR. Mitochondria in health and disease: perspectives on a new mitochondrial biology. Mol Aspects Med 2004;25:365-451.
- Neustadt J, Pieczenik SR. Medication-induced mitochondrial damage and disease. Mol Nutr Food Res 2008;52:780-8.
- Lee HC, Wei YH. Mitochondria and aging. Adv Exp Med Biol 2012;942:311-27.
- Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene 2006;25:4647-62.
- Hwang MS, Rohlena J, Dong LF, Neuzil J, Grimm S. Powerhouse down: Complex II dissociation in the respiratory chain. Mitochondrion 2014;19 Pt A:20-8.
- Ralph SJ, Moreno-Sánchez R, Neuzil J, Rodríguez-Enríquez S. Inhibitors of succinate: quinone reductase/Complex II regulate production of mitochondrial reactive oxygen species and protect normal cells from ischemic damage but induce specific cancer cell death. Pharm Res 2011;28:2695-730.
- Trifunovic A, Wredenberg A, Falkenberg M, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004;429:417-23.
- Trifunovic A, Hansson A, Wredenberg A, et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci U S A 2005;102:17993-8.
- Kujoth GC, Hiona A, Pugh TD, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005;309:481-4.
- Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD. ROS and the DNA damage response in cancer. Redox Biol 2019;25:101084.
- Szczesny B, Brunyanszki A, Olah G, Mitra S, Szabo C. Opposing roles of mitochondrial and nuclear PARP1 in the regulation of mitochondrial and nuclear DNA integrity: implications for the regulation of mitochondrial function. Nucleic Acids Res 2014;42:13161-73.
- Hocsak E, Szabo V, Kalman N, et al. PARP inhibition protects mitochondria and reduces ROS production via PARP-1-ATF4-MKP-1-MAPK retrograde pathway. Free Radic Biol Med 2017;108:770-84.
Clinical Topics: Cardio-Oncology, Dyslipidemia, Prevention, Lipid Metabolism, Stress
Keywords: Cardiotoxicity, Reactive Oxygen Species, Electron Transport, DNA, Mitochondrial, Telomere, Histones, Protons, Chromatin, Oxidative Phosphorylation, Activating Transcription Factor 4, Hydrogen Peroxide, Proton Pumps, Telomere Shortening, Mitochondrial Membranes, DNA Breaks, Double-Stranded, Proton-Motive Force, Dual Specificity Phosphatase 1, Citric Acid Cycle, Glycolysis, Paclitaxel, Aging, Premature, Cardiovascular Agents, Electrons, Zinc, Risk Factors, Xenobiotics, Membrane Proteins, Cardiovascular Diseases, Prevalence, Neoplasms, Mitochondrial Proton-Translocating ATPases, Mitochondria, Flavin-Adenine Dinucleotide, DNA Repair, Adenosine Triphosphate, Doxorubicin, Mutation, Energy Transfer, Inflammation, Oxidative Stress, Hydrogen-Ion Concentration, Cohort Studies
< Back to Listings