Written on behalf of the Sanger Heart and Vascular Education Group
A 60-year-old obese male patient with obstructive sleep apnea on continuous positive airway pressure presented to your office with worsening dyspnea and lower extremity edema. An echocardiogram was ordered showing a dilated right heart with pulmonary artery systolic pressure >60 mmHg.
When thinking about pulmonary arterial hypertension (PAH), which of the following statements is true?
Show Answer
The correct answer is: A. PAH is inherited in less than 10% of cases.
PAH is inherited in less than 10% of cases. Mutations in two genes in the transforming growth factor beta receptor pathway BMPR2 and activin-like kinase 1 have been implicated in the pathogenesis of familial PAH. BMPR2 modulates vascular cell growth.
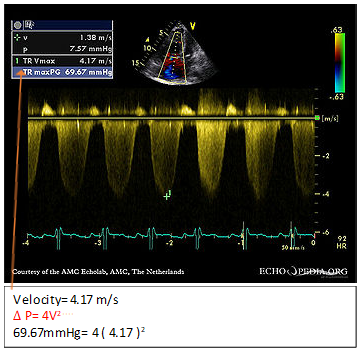
Regarding answer B, the spectral Doppler profile of tricuspid regurgitation is too weak or insufficient to measure the right ventricular (RV) to right atrial pressure gradient in approximately 10-25% of patients with PAH referred for evaluation. When this problem is encountered, a right heart catherization should be considered, especially if other features are present such as RV dilation/dysfunction. RV systolic pressure is estimated in the echocardiography laboratory by the following formula: Δ P = 4V2. The technician will measure the velocity of flow across the tricuspid valve to give an estimate of the right heart pressure (Figure 1).
Figure 1
Figure 1
Regarding answer C, the term pulmonary hypertension refers to the presence of abnormally high pulmonary vascular pressure. PAH is a category of pulmonary hypertension; the two terms are not synonymous. The conventional definition of PAH used in clinical studies includes an mPAP of greater than 20 mmHg at rest in the setting of a normal pulmonary arterial wedge pressure of 15 mmHg or less with a pulmonary vascular resistance greater than 3 Wood units. This hemodynamic definition has subsequently been applied in enrollment requirements in virtually every randomized clinical treatment trial, along with additional criteria including functional classification and 6-minute walk test to ensure that a relatively advanced stage of disease was studied.
Regarding answer D, mortality has never been proven by any trial. Measures of functional capacity and duration of the 6-minute walk test do improve.
Regarding answer E, the enthusiasm for the use of calcium channel blockers in idiopathic PAH dates back to 1992 with the publication of a study that demonstrated 95% 5-year survival in a very select group of patients with idiopathic PAH who exhibited an acute vasodilator response to calcium channel blockers. The current consensus definition of a response is now defined as a fall in mPAP of ≥10 mmHg to an mPAP ≤40 mmHg with an unchanged or increased cardiac output. Although many experts also perform vasodilator testing on patients with associated forms of PAH, true responders are very uncommon. Patients who meet these criteria may be treated with calcium channel blockers and should be followed closely for both safety and efficacy of calcium channel blocker therapy. If a patient who meets the definition of an acute response does not improve to functional class I or II on channel blocker therapy, the patient should not be considered a chronic responder, and alternative or additional PAH therapy should be instituted. Long-acting nifedipine, diltiazem, or amlodipine are the most commonly used calcium channel blockers. Due to its potential negative inotropic effects, verapamil should be avoided.
References
McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol 2009;53:1573-619.