A 17-Year Old Hockey Player With Aortic Abnormalities: Dissecting the Decisions Affecting Aortopathies and Sport Participation
A 17-year-old male competitive hockey player presents for evaluation after detection of an enlarged aorta. He underwent screening for aortic disease because his father had an aortic dissection and emergent aortic surgery at 23 years old. Through genetic testing, his father was subsequently found to have an ACTA2 mutation.
A transthoracic echocardiogram was performed demonstrating a tricuspid aortic valve without stenosis or regurgitation, normal left ventricular dimensions, left ventricular ejection fraction 60% and an aortic root diameter 3.47 cm measured at the sinus of Valsalva (patient height 172 cm, weight 67 kg, calculated Z score 2.28). Imaging of the aorta was additionally notable for a loss of the normal aortic contour and no discernible sinotubular junction. The ascending aorta was enlarged and measured 3.93 cm.
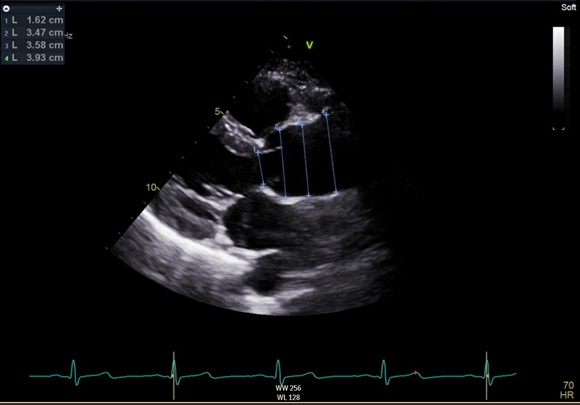
Figure 1
A chest magnetic resonance angiography (MRA) performed 15 months prior showed mild dilation of the aortic root (3.3 cm x 3.3 cm), measured at the sinus of Valsalva, and mild dilation of the ascending aorta (3.7 cm x 3.4 cm).
On physical examination, there were no physical stigmata to suggest a systemic connective tissue disorder. He was asymptomatic and reported no chest discomfort or shortness of breath at rest or with exertion.
What is the next step in the evaluation of this patient and what are your recommendations for sports participation?
Show Answer
The correct answer is: A. Order genetic testing and recommend against participation in hockey and other collision sports given the increased Z score and family history of aortic dissection.
The patient was referred for genetic testing and a genetic aortopathy panel was ordered that revealed an ACTA2 mutation, consistent with his father's mutation, and confirming a diagnosis of familial thoracic aortic aneurysm and dissection syndrome (TAAD). He was advised against participation in competitive hockey and other collision sports. Given his family history of aortic dissection, diagnosed genetic aortopathy and already enlarged aorta, he should not engage in competitive contact sports (hence, answers B-E are incorrect).
Genetic Aortopathies
Genetic aortopathies can be broadly divided into three categories. The first category includes aortopathies associated with syndromic disorders, such as Marfan Syndrome (MFS) or Loeys-Dietz Syndrome (LDS), where aortic aneurysms are one component of a systemic connective tissue disease. The second category includes the non-syndromic aneurysmal disorders – specifically familial TAAD – where the connective tissue pathology appears to be confined to the aorta. The third category includes aortopathies associated with congenital heart disease, typically a bicuspid aortic valve.
MFS is caused by a mutation in the gene encoding Fibrillin-1 (FBN-1). It is inherited in an autosomal dominant fashion but 25% of cases do not have identifiable family history and arise due to spontaneous mutations. Characteristic physical features include tall stature, thin frame, disproportionately long extremities, pectus deformities, scoliosis, stretch marks and ophthalmic disease, most notably ectopia lentis. The revised Ghent nosology delineate the criteria needed to confirm the diagnosis.1 For patients with MFS, echocardiogram studies at diagnosis and 6 months thereafter are recommended to establish the initial aortic size and rate of progression of the aortic dilation.2 Thereafter, an annual echocardiogram is recommended if the aortic diameter is <4.5 cm or more frequently if the aortic diameter is larger, if there is rapid progression of aortic size, or if there is severe valvular regurgitation. Surgery is recommended for all patients if the aortic diameter is at least 5.0 cm, if the aortic diameter is between 4.5-5.0 cm and an aortic valve-sparing operation is available, if the rate of aortic growth is at least 5 mm per year, if there is a family history of dissection at an aortic diameter <5 cm, or if there is severe mitral regurgitation (MR) or a need for another cardiac surgery.3
LDS is also an autosomal dominant systemic connective tissue disorder with four dominant genetic subtypes that include mutations in TGFBR1, TGFBR2, TGFB2 or SMAD3. Characteristic features include widespread arterial involvement and tortuosity, craniofacial abnormalities (widely-spaced eyes, cleft palate, bifid uvula, skull malformations), scoliosis, pectus deformities, translucent skin and easy bruising. Similar to MFS, patients with LDS require an echocardiogram at diagnosis and 6 months thereafter to establish the initial aortic size and rate of progression of aortic dilation. Annual echocardiograms are recommended, with more frequent imaging in the presence of rapid progression of aortic size, severe valvular regurgitation, a family history of dissection, or severe craniofacial features. Yearly whole body MRA from the cerebrovascular circulation to the pelvis is recommended.2 Importantly, thoracic aortic aneurysms have a more aggressive natural course in patients with LDS compared to those observed in MFS.4 Surgical recommendations are therefore more heterogenous and individualized than for MFS. Surgery is recommended for an aortic diameter of at least 4.2 cm by echocardiography (internal diameter) or 4.4 cm by computed tomography (CT)/MRA (external diameter)2 or at any aortic dimension if an aortic valve sparing operation is available. Surgery for LDS is also recommended if the rate of aortic growth is at least 5 mm per year, if there is a family history of dissection, or in the presence of severe aortic regurgitation (AR) or MR.2 In young children, specific prominent craniofacial features are associated with more severe aortic disease and in such cases, prophylactic surgery can allow for the placement of a graft of sufficient size to accommodate growth once the aortic diameter surpasses the 99th percentile for age and aortic valve annulus is 1.8 to 2.0 cm.2
Familial TAAD is a heterogeneous group of non-syndromic disorders characterized by aortic aneurysmal disease in multiple family members. Affected individuals do not meet criteria for a systemic connective tissue disorder. These genetic disorders are inherited in an autosomal dominant fashion, however there is significant variability in expressivity and penetrance.5 The most commonly identified mutations involve ACTA2 (10-14%) as well as MYH11, MYLK, PRKG1 and TGFBR2.5 The 2010 American Heart Association (AHA)/American College of Cardiology (ACC) guidelines recommend that first-degree relatives of individuals with an unexplained aortic aneurysm or dissection should undergo aortic imaging (Class I, Level of Evidence B).2 In addition, if a mutant gene associated with TAAD is identified in a patient with aneurysmal disease, then first-degree relatives should undergo genetic counseling, testing and serial aortic imaging if the testing also identifies the mutation (Class I, Level of Evidence C), as was the case for this specific hockey player.2 For patients with no known aortic pathology but a confirmed genetic mutation known to be associated with aortic dissections, the 2010 guidelines suggest complete aortic imaging at diagnosis and again after 6 months to evaluate for aortic growth or development of pathology.2
Sport Participation Recommendations
Prior recommendations from the 36th Bethesda Conference (2005) suggest that athletes with a known genetic aortopathy or "unequivocal aortic root enlargement" (defined as >40 mm in adults, >2 standard deviations above the mean for body surface area (BSA) in children and adolescents or a Z score of >2), irrespective of the presence or absence of a known genetic aortopathy, should only participate in low intensity and non-contact competitive sports.6
In 2015, the AHA/ ACC Task Force 7 updated these recommendations to differentiate athletes with a known aortopathy from athletes without a known aortopathy but with enlarged aortic dimensions. In these updated Task Force guidelines, serial aortic imaging is recommended for athletes without a detectable genetic aortopathy but sport participation is not restricted unless clinical concerns arise regarding aortic dimensions.
An important element to the 2015 recommendations is that any athlete with enlarged aortic dimensions should be counseled in a shared decision construct regarding the potential risks of further training and competition.7 The discussions should include the athlete as well as parents (if applicable) and coaches/trainers. Embedded within the discussion should be the recognition that the recommendations are based on a low level of evidence (Level of Evidence C) and that uncertainty exists regarding the extent to which sport participation itself leads to aortic remodeling and dilation. For example, Churchill et al. demonstrated increased aortic dimensions in a significant portion of masters endurance athletes (runners and rowers) engaged in long-term intensive training.8 Separating attributes of possible athletic cardiovascular remodeling from clear pathology that poses a risk of an aortic emergency can be exceptionally challenging in those athletes with increased aortic dimensions but without a defined genetic aortopathy.
The table below illustrates recommendations adapted from those guidelines.
| Patient Population | Recommendations | Class/Level of Evidence |
| Athletes with unexplained aneurysm, familial TAAD or a known pathogenic mutation leading to a familial TAAD (ACTA2, MYH11, FBN1, TGFBR1, TGFBR2, MLCK, SMAD3, TGFB2 and others). | Surveillance Echo and (depending on the diagnosis) MRA or CT every 6-12 months to monitor for progression of aortic or branch vessel disease. Participation Reasonable to participate in low static, low dynamic (class IA; definitions in Task Force 1)9 competitive sports if they do not have concerning features.* |
I/C IIa/C |
| Athletes with mildly dilated aortic dimensions (Z scores 2-2.5 or aortic root diameters 40-41 mm in tall men or 36-38 mm in tall women) and no features of MFS, LDS, familial TAAD or a bicuspid aortic valve. | Surveillance Echo or MRA every 6-12 months with frequency depending on the aortic size and stability of measurement. Participation Participation in all competitive athletics may be considered after a comprehensive evaluation for an underlying genetic condition associated with aortopathy is performed. This may include analysis for mutations in FBN1and other genes associated with aortopathies in certain circumstances. Avoidance of intense weight training may be considered. |
I/C IIb/C IIb/C |
| Athletes with MFS, familial TAAD, LDS, unexplained aortic aneurysm, vascular Ehlers-Danlos syndrome or a related aortic aneurysm disorder. | Participation Should not participate in any competitive sports that involve intense physical exertion or the potential for bodily collision. |
III/C |
| Athletes with MFS. | Surveillance Echo (and in some instances MRA or CT) measurement of the aortic root dimension every 6-12 months, depending on aortic size. Participation Reasonable to participate in low-moderate static/low dynamic competitive sports (classes IA and IIA)9 if they do not have any concerning features.** |
I/C IIa/C |
| Athletes with LDS or vascular Ehlers-Danlos. | Participation Reasonable to participate in low static, low dynamic sports (class IA)9 if they do not have any concerning features.*** |
IIa/C |
**Aortic root dilation (i.e.: z score >2 or aortic root diameter >40 mm or >2 SD from the mean relative to BSA for children and adolescents <15 years old), moderate to severe MR, family history of aortic dissection at an aortic diameter <50 mm, left ventricular systolic dysfunction (ejection fraction <40%)
***Aortic enlargement (z score >2) or dissection or branch vessel enlargement, moderate to severe MR, extracardiac organ involvement that makes participation hazardous
Follow Up
The patient has discontinued participation in competitive hockey. He was advised to avoid strenuous isometric exercise but in accordance with recommendations continues to engage in recreational dynamic sports and exercise. He takes an angiotensin receptor blocker daily and has ongoing surveillance aortic imaging every 6 months.
References
- Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010;47:476–85.
- Hiratzka LF, Bakris GL, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2010;121:e266-369.
- Erbel R, Aboyans V, Boileau C, et al. 2014 ESC guidelines on the diagnosis and treatment of aortic diseases: document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J 2014;35:2873–926.
- Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 2005;37:275–81.
- Goyal A, Keramati AR, Czarny MJ, Resar JR, Mani A. The genetics of aortopathies in clinical cardiology. Clin Med Insights Cardiol 2017;11:1179546817709787.
- Maron BJ, Ackerman MJ, Nishimura RA, Pyeritz RE, Towbin JA, Udelson JE. Task Force 4: HCM and other cardiomyopathies, mitral valve prolapse, myocarditis, and Marfan syndrome. J Am Coll Cardiol 2005;45:1340–5.
- Braverman AC, Harris KM, Kovacs RJ, Maron BJ. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 7: aortic diseases, including Marfan Syndrome: a scientific statement from the American Heart Association and American College of Card. J Am Coll Cardiol 2015;662398-2405.
- Churchill TW, Groezinger E, Kim JH, et al. Association of ascending aortic dilatation and long-term endurance exercise among older masters-level athletes. JAMA Cardiol 2020;5:522–31.
- Levine BD, Baggish AL, Kovacs RJ, Link MS, Maron MS, Mitchell JH. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 1: classification of sports: dynamic, static, and impact: a scientific statement from the American Heart Association and American College of Cardiology. J Am Coll Cardiol 2015;66:2350-55.