A previously healthy 13-year-old girl presents to the emergency department (ED) after an episode of syncope. Her older sister reported that, after sitting up in bed in the morning, the patient made a noise and then her eyes rolled back, and extremities stiffened. She was unresponsive for approximately 30 sec and then recovered to baseline in a few minutes. She has previously had brief episodes of palpitations but no syncope. There is no significant family medical history, and she is not taking any medications.
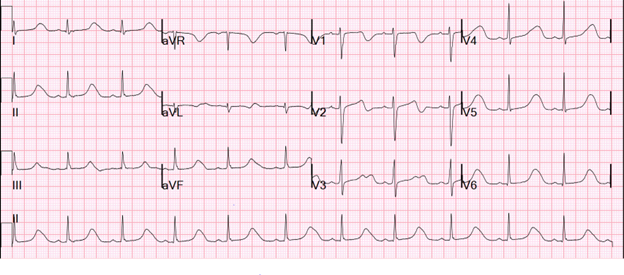
In the ED, she is alert and active. Her vital signs include temperature 36.9°C (98.4°F), heart rate 66 bpm, blood pressure 110/82 mm Hg, and oxygen saturation 99%. Laboratory study findings are notable for blood count and electrolyte levels within the reference ranges. A 12-lead electrocardiogram (ECG) (Image 1) and a cardiology consult are obtained.
Image 1
The correct answer is: C. Promptly initiate beta-blocker therapy.
The 12-lead ECG (Image 1) has findings of a markedly prolonged QT interval at 604 msec (QTc 634 msec) and abnormal T-wave morphology. Together with syncope, these findings strongly suggest congenital long QT syndrome (LQTS) and an arrhythmogenic cause of this patient's syncope.1,2 Per guidelines, beta-blocker therapy is recommended for patients suspected of having or confirmed to have LQTS who are symptomatic with syncope or have documented ventricular tachycardia/fibrillation.3
Although she might require an ICD in the future, she did not experience cardiac arrest or require cardiopulmonary resuscitation and was not taking beta-blocker therapy when she had her syncopal event. ICD implantation is usually reserved for survivors of cardiac arrest (Class 1) or those who experience recurrent events while taking beta-blocker therapy (Class 2a).3
Discharge with outpatient follow-up without beta-blocker therapy would not be appropriate because she had risk factors for sudden cardiac death (syncope and marked QT prolongation). Although QT prolongation may be seen following syncope from a variety of causes, the magnitude of her QT prolongation made it unlikely to be a benign secondary phenomenon. In addition, she did not have any other obvious secondary cause of QT prolongation.
She was started on beta-blocker therapy with nadolol.4 Genetic testing had positive findings of a pathogenic mutation in sodium voltage-gated channel alpha subunit 5 (SCN5A), consistent with a diagnosis of congenital LQTS type 3. Given her persistently prolonged QTc interval on ECG and to reduce possibility of arrhythmic events, mexiletine was prescribed in addition to nadolol, which led to a shortening of the QTc to 500 msec.5 Subsequently, she had documented nonsustained polymorphic ventricular tachycardia on a Holter monitor. After shared decision-making with her family, a dual-chamber transvenous ICD was implanted for protection and atrial pacing to further shorten her QT interval.6 She remained free of symptoms and arrhythmia on nadolol and mexiletine therapy, and her QTc interval was 440-460 msec with atrial pacing at 70 bpm.
References
- Schwartz PJ, Ackerman MJ. The long QT syndrome: a transatlantic clinical approach to diagnosis and therapy. Eur Heart J 2013;34:3109-16.
- Wilde AAM, Amin AS, Postema PG. Diagnosis, management and therapeutic strategies for congenital long QT syndrome. Heart 2022;108:332-8.
- Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013;10:1932-63.
- Ackerman MJ, Priori SG, Dubin AM, et al. Beta-blocker therapy for long QT syndrome and catecholaminergic polymorphic ventricular tachycardia: are all beta-blockers equivalent? Heart Rhythm 2017;14:e41-e44.
- Mazzanti A, Maragna R, Faragli A, et al. Gene-specific therapy with mexiletine reduces arrhythmic events in patients with long QT syndrome type 3. J Am Coll Cardiol 2016;67:1053-8.
- Webster G. Revisiting atrial pacing in the long QT genotype era. J Cardiovasc Electrophysiol 2021;32:790-1.