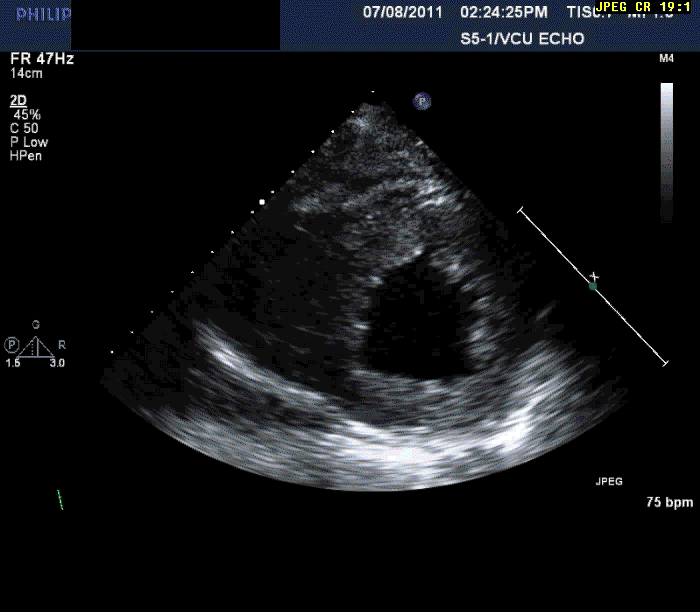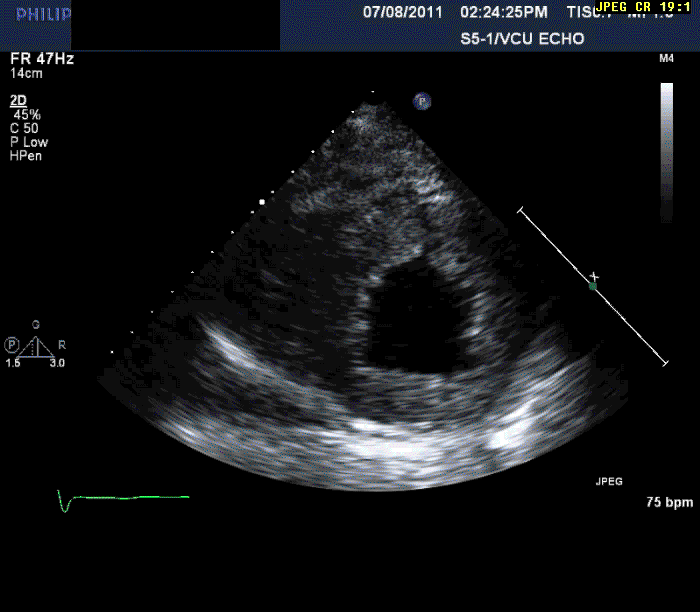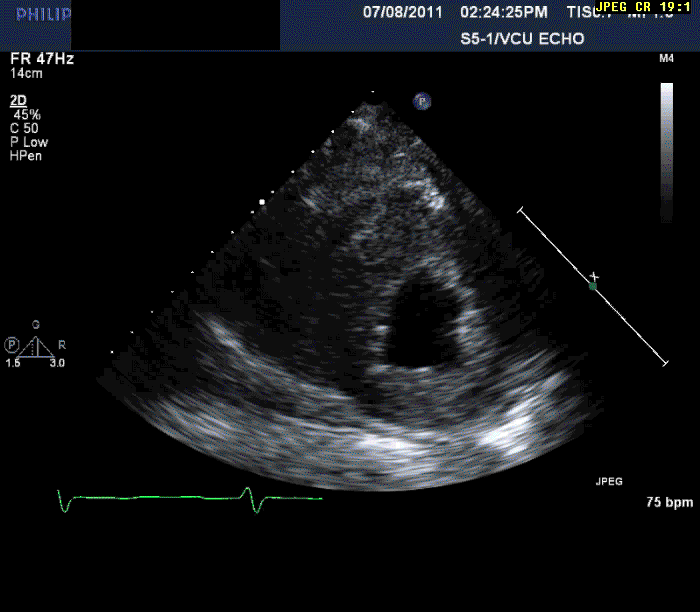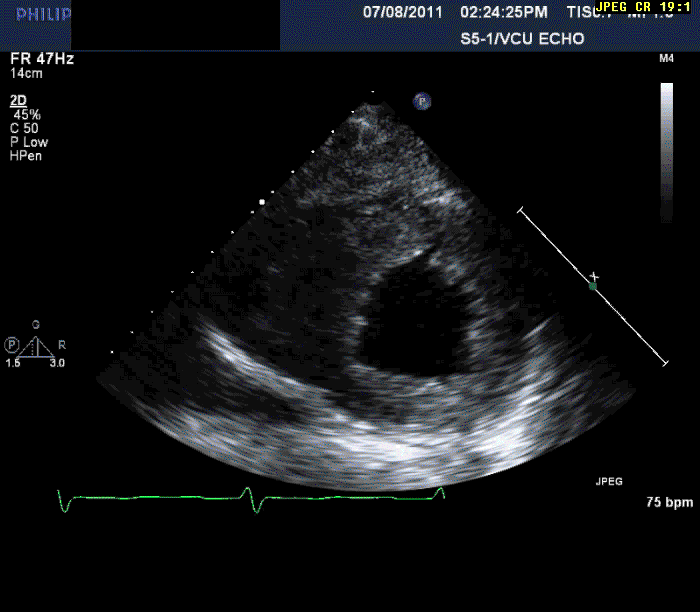
The patient is a 56-year-old male who presents with shortness of breath. The patient had been in his usual state of health until approximately three months prior to evaluation. At that time, he developed progressive shortness of breath on exertion, lower extremity edema, and in the last month three pillow orthopnea with PND 2-3 times per week. He denied any ongoing chest pain, pressure, tightness, or squeezing.
Past Medical History: Notable for no prior significant cardiac history, specifically, no MI or revascularization. Increased cholesterol controlled with diet.
Medications:
Intermittent non-steroidal use for aches and pains.
Physical Examination: BP 95/60 mmHg. Heart rate 65 BPM. Respiratory rate 22. HEENT: JVD to the neck at 45º Lungs: bibasilar rales Cardiac: regular rate and rhythm. No murmurs. (+) S4 Abdomen: mildly obese, bowel sounds present, nontender, non-extended Peripheral pulses: 2+ without bruits Extremities: 1-2+ bilateral, symmetrical lower extremity edema.
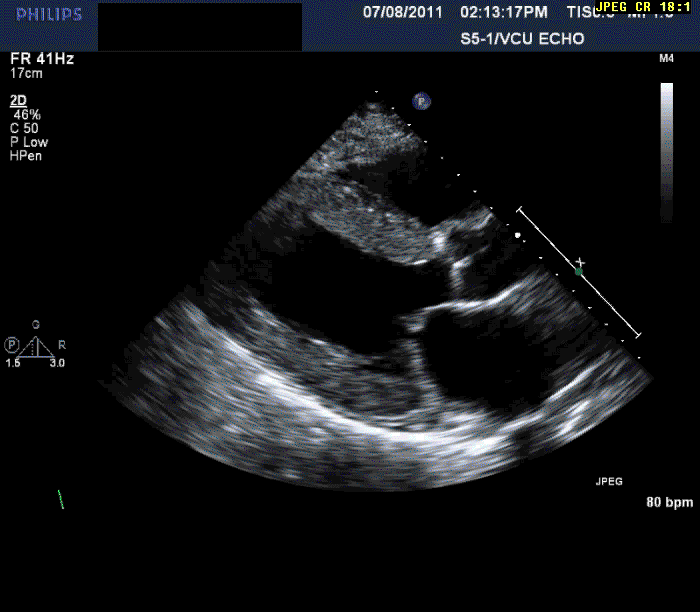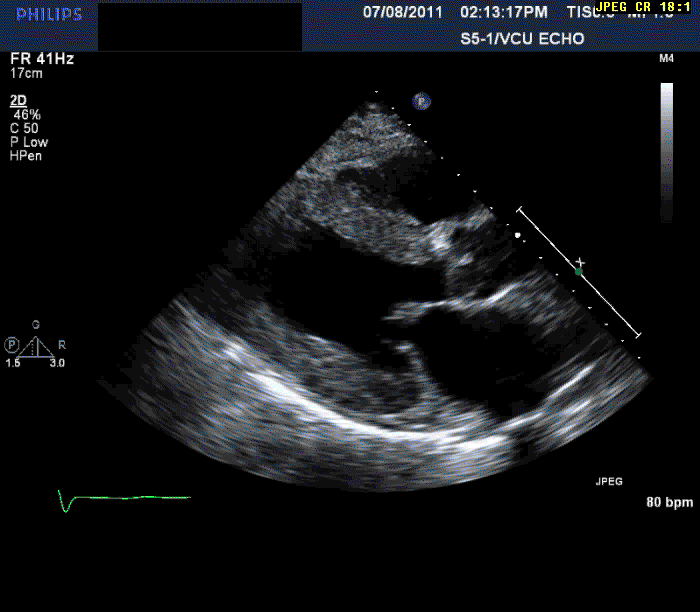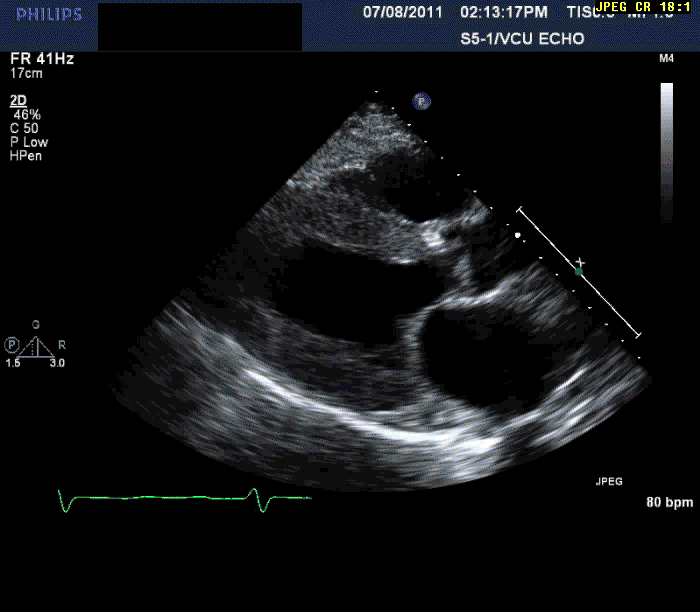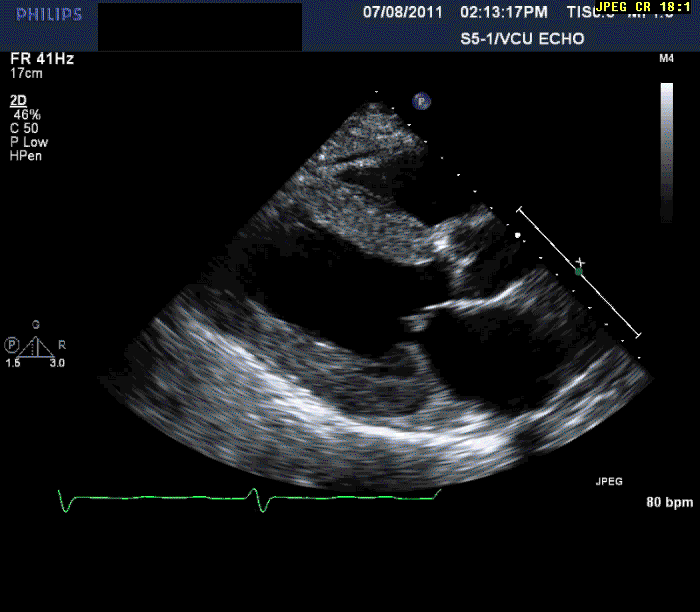
Normal LV cavity size (LVDD 4.5 cm, septum 1.5 cm, posterior wall 1.5 cm) with severe left ventricular hypertrophy (LV mass=180 gm/mm²) and severe systolic dysfunction (LVEF 40%)
Mild mitral regurgitation
Mild tricuspid regurgitation
Normal right ventricular size with moderately reduced function.
Given this initial presentation, what would be your next diagnostic choice?
Show Answer
The correct answer is: 4. Cardiac MRI
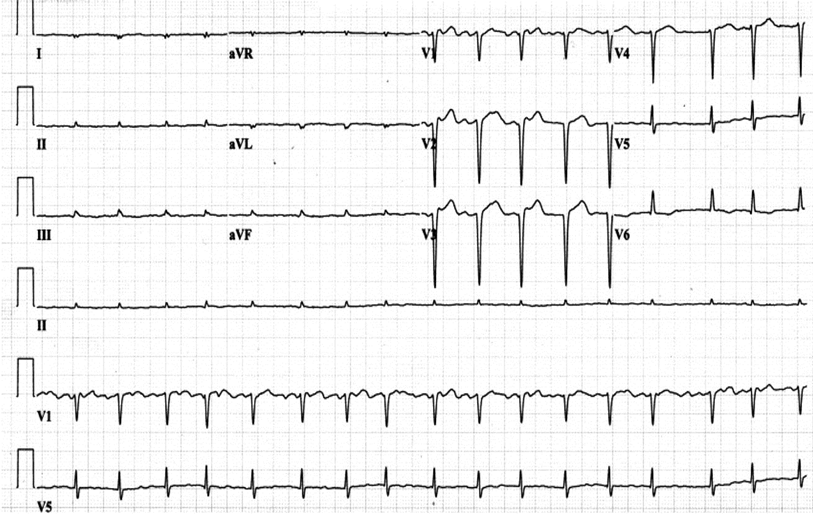
The patient's initial presentation is consistent with progressive cardiomyopathy of unknown cause. The echocardiogram is consistent with significant LVH. However, in the absence of a significant history of hypertension, other types of cardiomyopathy should be considered. The low voltage on the ECG could be related to prior MI but is also suggestive of an infiltrative disease. Cardiac MRI offers a unique diagnostic test which gives the ability to differentiate many types of cardiomyopathy.
Amyloidosis is a clinical disorder caused by extracellular deposition of pathologic insoluble abnormal fibrillar protein in organs and tissues. Cardiac amyloidosis can occur in systemic amyloidosis, in which amyloid deposits in organs, blood vessel walls, and connective tissues, or when it is limited to the heart alone.
Echocardiography can often offer clues to the diagnosis, such as in this case. The most common echocardiographic feature is left ventricular wall thickening, particularly in the absence of hypertension.1 Although frequently referred to as hypertrophy, increased wall thickness is due to infiltration, not myocyte hypertrophy. Increased wall thickness has a poor specificity for amyloidosis because of its frequent occurrence with other conditions; however, when it is seen in the absence of a significant hypertension history, as was seen in this case, cardiac amyloidosis should be considered. Increased echogenicity of the myocardium, particularly with a granular or "sparkling" appearance, has been reported as a diagnostic criterion2 but appears to have lower specificity in clinical practice.
ECG findings present in patients with cardiac amyloidosis include low voltage, which in one study was present in 46% of patients, and a pseudo-infarct pattern in 47% of patients, with both present in 25% of patients.3
Cardiac magnetic resonance imaging (cMRI) has gained an important role in the diagnosis of cardiac amyloidosis.4 Delayed post-gadolinium enhancement images reveal the characteristic diffuse subendocardial enhancement differentiating cardiac amyloidosis from other cardiomyopathies. The degree of enhancement depends on the ventricular mass and global ventricular systolic function.4 The overall accuracy for diagnosing cardiac amyloidosis has an accuracy of 97%.4 Although imaging is useful, histopathology is required, with further genetic testing for definitive diagnosis.
Both troponin and the natriuretic peptides have been found to be elevated in infiltrative cardiomyopathies, including amyloid. Myonecrosis and small-vessel ischemia due to amyloid deposit lead to an increase in cardiac troponins,5 whereas diastolic dysfunction and increased genetic expression of natriuretic peptide genes in the amyloid infiltrated ventricles lead to an increase in B-type natriuretic peptide (BNP) levels.6 Both markers of myocardial damage (Troponin T and I) and myocardial stretch (BNP and N-terminal (NT)-proBNP) provide prognostic information.
In one of the first studies to describe the role of troponin, Dispenzieri et al.5 found that in 261 newly diagnosed patients with cardiac amyloidosis, 163 (62%) had detectable amounts of cTnT (≥0.01 μg/L) and 230 (88%) had detectable cTnI (≥0.03 μg/L). A total of 59 (23%) had what were considered "large" values of cTnT (≥0.1 μg/L), and 140 (54%) had high cTnI (≥0.1 μg/L). Detectable cTnI and cTnT were associated with a median survival of only 6 or 8 months (respectively), compared with 22 or 21 months (respectively) in those without any detectable cTnI/T. Elevation in cTnI/T was the most important independent prognostic variable, outweighing echocardiographic variables.5
Naturietic peptides have also be shown to confer prognostic significance. In one large study of 810 patients with newly diagnosed amyloidosis, a prognostic model that was developed found that cTnT and NT-ProBNP were among the four independent prognostic variables for survival.7 NT-proBNP rapidly decreases with therapy and therefore may be a useful marker of response.8
In contrast to the consistent predictive value of cTn for predicting outcomes, a more recent study that incorporated newer echocardiographic measurements such as strain rate imaging, found that BNP and cTnT were univariate predictors, but only BNP was a multivariate predictor of mortality.1 cTnT correlated with heart failure class, ejection time, and septal strain of the anteroseptal segment, which were stronger predictors of mortality.
References
Bellavia D, Pellikka PA, Al-Zahrani GB, et al. Independent predictors of survival in primary systemic (Al) amyloidosis, including cardiac biomarkers and left ventricular strain imaging: an observational cohort study. J Am Soc Echocardiogr 2010;23:643-52.
Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol 2004; 43:410–415.
Murtagh B, Hammill SC, Gertz MA, et al. Electrocardiographic findings in primary systemic amyloidosis and biopsy-proven cardiac involvement. Am J Cardiol 2005; 95:535–537.
Maceira AM, Joshi J, Prasad SK, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2005; 111:186–193.
Dispenzieri A, Kyle RA, Gertz MA, et al. Survival in patients with primary systemic amyloidosis and raised serum cardiac troponins. Lancet 2003; 361:1787–1789.
Takemura G, Takatsu Y, Doyama K, et al. Expression of atrial and brain natriuretic peptides and their genes in hearts of patients with cardiac amyloidosis. J Am Coll Cardiol 1998; 31:754–765.
Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol 2012;30:989-95.
Palladini G, Campana C., Klersy C., et al. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation 2003;107:2440–2445.