Cover Story | Cardiac Amyloidosis: Ready For Prime Time
A recently published case study describes a 72-year-old man with a history of hyperlipidemia and palpitations presenting for ablation of supraventricular tachycardia.1
His history is also notable for a knee replacement, two shoulder surgeries and bilateral carpal tunnel syndrome. Cardiac magnetic resonance (CMR) imaging after the procedure showed prominent thickening of the interventricular septum and a spontaneous intramyocardial hemorrhage.
Further testing revealed extensive global late gadolinium enhancement which suggested amyloid infiltration, normal serum and urine protein electrophoresis, a positive fat pad biopsy for amyloid, a positive pyrophosphate (PYP) nuclear scan, and a wild-type transthyretin TTR gene.
The diagnosis of wild-type transthyretin amyloid cardiomyopathy (ATTR-CM) was made and the patient started on tafamidis (Click here for an overview of amyloidosis and the nomenclature).
"Everything about this case is typical and common except for the spontaneous intramyocardial hemorrhage, which is extraordinarily rare," says Ronald Witteles, MD, FACC, co-director of the Stanford Amyloid Center at Stanford University and associate editor of JACC: CardioOncology.
"But cardiac amyloidosis is by no means rare and if you look in the right populations, you'll find it again and again," he adds.
With new advances in diagnosis and treatment, all that's needed now to see real progress in patient outcomes is a greater awareness of the disease and its impact on the heart, along with an understanding of its diagnosis and the available treatment options.
"Not a Zebra"
 Witteles, R.M. et al. J Am Coll Cardiol HF. 2019;7(8):709-16. Click the image above for a larger view.
Witteles, R.M. et al. J Am Coll Cardiol HF. 2019;7(8):709-16. Click the image above for a larger view.
Cardiac amyloidosis is not a new disease. It is, however, a disease with new advances that are driving it out of the shadows and into the mainstream. For Mathew Maurer, MD, FACC, who has been studying cardiac amyloidosis for 15 years, it's an exciting time.
"In a very short time, cardiac amyloidosis has gone from a rare, underdiagnosed and difficult-to-diagnose disease with no specific treatment, to being much more common than people thought, easy to diagnose and treatable," Maurer says.
A trio of simultaneous areas of progress have propelled the field to new levels: a better understanding of the epidemiology, an accurate noninvasive means of diagnosis, and an approved therapy, according to a recent American Heart Association (AHA) Scientific Statement on cardiac amyloidosis.2
"These seminal advances have really shown us just in the last few years that cardiac amyloidosis is not the zebra we once thought it was," says Maurer who was the vice-chair on the AHA statement and is director of the Cardiac Amyloidosis Program at New York-Presbyterian Hospital/Columbia University Irving Medical Center.
Not only is the prevalence of cardiac amyloidosis higher than once thought, its impact on the heart is greater than once thought. The disease is increasingly recognized as an important and underdiagnosed cause of heart failure, particularly heart failure with preserved ejection fraction (HFpEF).
Increased ventricular wall thickness caused by amyloid fibril deposition is a prominent characteristic of ATTR-CM and can lead to ventricular stiffening and left ventricular (LV) diastolic dysfunction. It has also been fingered as an underlying cause of conduction abnormalities and atrial and ventricular arrhythmias.
In one small study, 13.3% of White adults 60 years and older admitted with HFpEF and LV wall thickness (12 mm) were found to have ATTR amyloidosis.3 ATTR amyloidosis also can be seen in one of six patients with severe calcific aortic stenosis undergoing TAVR.4
"It turns out that probably 1 or 2% of people over the age of 75 in general medical practices likely have cardiac amyloidosis, with the biggest driver just being that people are living longer. This is predominantly a disease of older age," says Maurer. There is also a male preponderance for reasons that are unclear.
In addition to the more common "wild-type" (nonhereditary) form, in the U.S., the hereditary form of ATTR-CM is predominantly a disease found in Blacks, a possible biological explanation for the disparate HF outcomes seen in this population.5 The frequency of the disease-causing mutation (TTR V122I) is about 1:29.6
"If you went to a newborn nursery today and sequenced their TTR gene, somewhere around one in 25 to 30 of babies who are Black in that nursey – between 3 and 4% – would have a variant in the TTR gene called Val121Ile," says Maurer. Thankfully, carrying the mutation does not guarantee the eventual development of the disease.
In a study just published in JACC: CardioOncology, a secondary diagnosis of amyloidosis (by ICD coding) was found in 2,845 (or 0.18%) of >1.5 million HF hospitalizations included in the National Readmissions Database.
Matched to 8,515 HF hospitalizations without amyloidosis, the patients with amyloidosis were more likely to also have kidney disease (56% vs. 45%), malignancy (20% vs. 4%) and higher inpatient mortality (6% vs. 3%).
After adjustment, HF with amyloidosis increased the odds of in-hospital mortality by 46% and 30-day readmission by 17%.7
These findings have implications not just for patient care (the condition is still underdiagnosed and specific protocols are needed to improve outcomes), but also for hospitals who care for high proportions of patients with amyloidosis who may be disadvantaged under the Hospital Readmissions Reduction Program because HF etiology is not taken into account when assessing hospital performance.8
"When I give talks on cardiac amyloidosis, I tell cardiologists and internists, 'If you haven't seen a patient with amyloidosis in your practice in the last year, it's not because it wasn't there, it's because you missed it,'" says Witteles.
Who to Screen: Stick to the Basics + the Red Flags
 Witteles, R.M. et al. J Am Coll Cardiol HF. 2019;7(8):709-16. Click the image above for a larger view.
Witteles, R.M. et al. J Am Coll Cardiol HF. 2019;7(8):709-16. Click the image above for a larger view.
Definitive diagnosis of amyloidosis used to require endomyocardial tissue biopsy. The good news is that now biopsy is only done as a confirmatory measure, if at all. The better news is that screening for cardiac amyloidosis today is much easier than most cardiologists realize.
In a state-of-the-art review on screening for ATTR-CM in everyday practice in JACC: Heart Failure, a group of experts led by Witteles present evidence-based recommendations on who and how to screen (Figure 1).9
The paper stresses both the importance and the feasibility of screening in the wider cardiology community.
"We defined who to screen very simply: men over 65 years and women over 70 years who have heart failure and a thickened ventricle, defined as anything 14 mm or thicker. That's it! That alone should prompt screening," says Witteles.
"Beyond this basic recommendation, if a patient presents with one of our "red flag" signs or symptoms (Figure 2), screening should be considered even more," adds Witteles.
The red flags are clinical cues that should further heighten suspicion or alert clinicians to the possibility of ATTR-CM. For example, studies have shown that screening tissue from individuals undergoing surgery for carpal tunnel syndrome will reveal amyloid in about 10% of samples. For those undergoing a second procedure on the contralateral arm, the proportion goes up even more.
"When the red flags are added, it makes it even more likely the patient may have amyloidosis," says Witteles. "We stress, however, screening does not need to be restricted to the red flag clinical scenarios. The basic screening scenario is simple: older men and women with heart failure and increased wall thickness," stresses Witteles.
To this list of red flags, Debabrata Mukherjee, MD, FACC, would add rupture of the distal biceps tendon, citing a study in JAMA that found distal tendon rupture in 33.3% of patients with wild-type ATTR-CM, vs. 2.5% of patients with other causes of HF.10
He is in full agreement that cardiac amyloidosis should be considered in older patients with HF and LV hypertrophy.
"Heart failure with hypertension or coronary artery disease are far more common, but cardiac amyloidosis is not nearly as uncommon as people think. If you see someone with HF, particularly with preserved ejection fraction, and LV hypertrophy, you need to screen them," Mukherjee says.
A greater awareness of the epidemiology of cardiac amyloidosis can increase the chances of making the diagnosis in a timely fashion, notes Mukherjee, chief of cardiology at Texas Tech University and an associate editor of the ACC.org Editorial Team.
"We need to think about these things, because you can't find something you're not looking for," says Mukherjee. He notes he has diagnosed cardiac amyloidosis in several patients – and the most recent had HFpEF with LV hypertrophy and a history of unilateral carpal tunnel syndrome.
How to Screen: Blood Tests + Scan = Diagnosis
 Click the image above for a larger view.
Click the image above for a larger view.
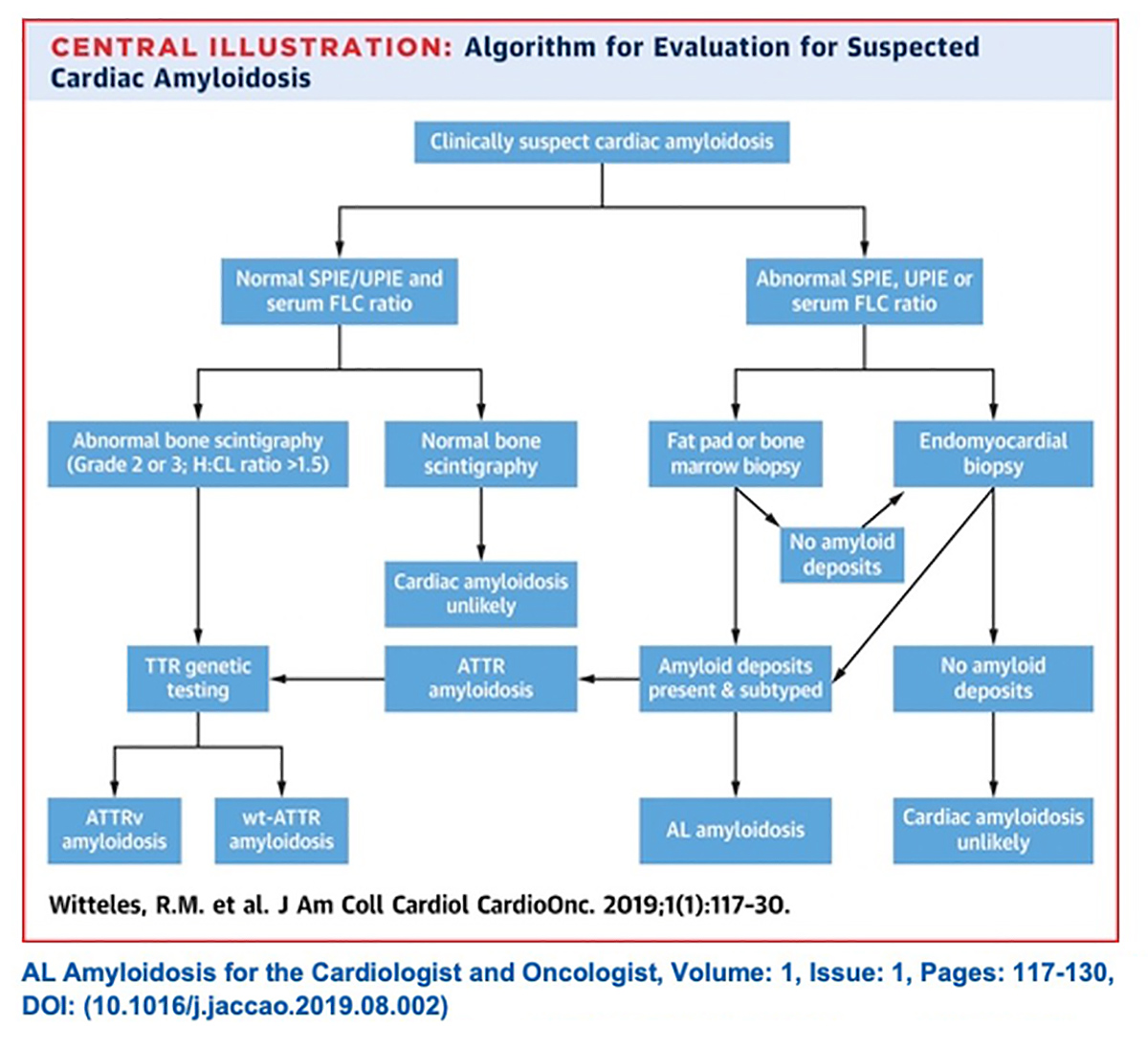
Once there is clinical suspicion for cardiac amyloidosis and a decision to screen has been made, it's essentially a two-step process to a noninvasive diagnosis of ATTR-CM: some lab work and a nuclear scan (Figure 3).
"Ideally the first step is to run three laboratory tests: serum free light chain assay, serum protein electrophoresis with immunofixation (SPIE) and urine protein electrophoresis with immunofixation," says Witteles.
He notes it's best to include the urine protein electrophoresis but not critical.
These two tests – serum free light chain assessment and SPIE – combined have a 99% sensitivity for identifying someone at risk for amyloid light chain (AL) amyloidosis. If the tests are normal, the next step is imaging to evaluate for ATTR amyloidosis.9
"The possibility of misdiagnosis underscores the importance of conducting the lab tests before any imaging," says Witteles. If the scan is negative, which is not uncommon because AL amyloid often won't be seen on a PYP scan, the diagnosis of AL amyloidosis could be missed.
Conversely, if the PYP is positive (which it is in about a quarter of AL amyloidosis cases) in the absence of lab test results, an incorrect diagnosis of ATTR amyloidosis could be made and the patient could be started on tafamidis, which is the wrong treatment, rather than chemotherapy or immunotherapy.
"Missing a diagnosis of AL amyloidosis is a catastrophe because it can be so rapidly progressive," says Witteles.
With lab results in hand, next up is imaging. Classic imaging features on echocardiography and CMR can be supportive of a diagnosis of infiltrative cardiomyopathy, but are not specific enough to diagnose cardiac amyloidosis or distinguish AL from ATTR. However, accumulating evidence now supports the use of bone scintigraphy for this purpose.
Scintigraphy with technetium (Tc)-labeled bisphosphonates localizes to TTR cardiac amyloid deposits, although the molecular basis is not clear. 99mTc-labeled 3,3-diphosphono-1,2-propanodicarboxylic acid (DPD), 99mTc-labeled PYP and 99mTc-labeled hydroxymethylene diphosphonate (HMDP) have all shown high sensitivity and specificity for imaging TTR amyloid.
The scan is done at rest and a planar image is acquired initially followed by a SPECT study.
Maurer says the key here is: a planar image plus SPECT study. "The SPECT study is needed to confirm the uptake is in the myocardium," he explains. On planar imaging, it is not possible to distinguish whether the uptake of PYP is in the LV or right ventricular chamber, or in the myocardium.
Once AL is ruled out from the absence of detectable monoclonal protein, the specificity of PYP bone scintigraphy for ATTR-CM approaches 100%, says Witteles.
In most cases, endomyocardial biopsy is only needed if a monoclonal protein is detected, or if imaging results are equivocal. Once a diagnosis of ATTR-CM is made, genetic testing can differentiate between the hereditary and wild-type forms of the disease.
Treatment of ATTR-CM
Therapy for ATTR-CM focuses mainly on the management of HF or arrhythmias, anticoagulation as indicated for atrial fibrillation/flutter (regardless of the CHA2DS2-VASc score), and disease-modifying therapy.
The only disease-modifying therapy for ATTR-CM approved by the U.S. Food and Drug Administration (FDA) is the TTR stabilizer tafamidis, and this currently represents standard of care. Others are in development, including the next-generation form of patisiran (TTR silencing agents that target hepatic synthesis), and acoramidis, a TTR stabilizer.
Patisiran and inotersen are FDA approved drugs that work by knocking down the liver's production of TTR by about 85%, according to Witteles.
"For the hereditary TTR-mediated polyneuropathy form of the disease, those two drugs were absolutely spectacular in their clinical trials. I'm not sure I've ever seen more impressive results, for patisiran in particular," he says.
The next generation forms of these two drugs are currently being tested for ATTR-CM in two Phase 3 clinical trials (HELIOS-B and CardioTTRansform).
Tafamidis: A First-in-Class Treatment
 Witteles, R.M. et al. J Am Coll Cardiol HF. 2019;7(8):709-16. Click the image above for a larger view.
Witteles, R.M. et al. J Am Coll Cardiol HF. 2019;7(8):709-16. Click the image above for a larger view.
Tafamidis in an orally active drug that binds to the thyroxine site of TTR, stabilizing it, and inhibiting or slowing the formation of amyloid fibrils. The drug received FDA approval in May 2019 for the treatment of ATTR-CM. The approval was based on the findings of the ATTR-ACT study, presented by Maurer and colleagues at the ESC Congress 2018.11
The trial evaluated the safety and efficacy of tafamidis (20 or 80 mg) in 441 patients with hereditary or wild-type ATTR and HF. In keeping with the groups most affected by this disease, the majority of participants were male (90%) and had wild-type ATTR (76%); mean age was 75 years; and 14% of participants were Black. Randomization was stratified according to genotype (wild-type or variant) and disease severity (NYHA class).
In the primary analysis, which hierarchically assessed all-cause mortality followed by rates of cardiovascular-related hospitalization, tafamidis treatment was associated with a significant 30% reduction in all-cause mortality after 30 months (29.5% vs. 42.9%; hazard ratio [HR], 0.70; p=0.026).
Cardiovascular hospitalization was reduced 32% with treatment (0.48 per year vs. 0.70; relative risk reduction 0.68; p<0.0001), and significant improvements seen in secondary endpoints related to quality of life and exercise tolerance. No significant safety concerns were detected and the drug was well tolerated.
"Tafamidis makes people live longer, so we're no longer in a situation of diagnosing people with something for which we have no treatment," says Maurer, principal investigator of ATTR-ACT.
It should be noted that in patients with NYHA class III disease at baseline, the rates of cardiovascular-related hospitalizations were higher among patients receiving tafamidis than among those receiving placebo. The mortality benefit of tafamidis appeared to be attenuated in this subgroup. Maurer shares some detailed outcomes that suggest tafamidis works better when treatment starts earlier in the course of the disease.
In trial participants who were in NYHA class I HF (<10% of participants), the mortality risk reduction was 64%. In class II (the majority of participants), it was 39%. In class III HF, it dropped to 16%. The trial was a watershed moment for the disease.
"Until now, we were treating the manifestations of cardiac amyloidosis. We thought these people had HFpEF or a restrictive cardiomyopathy, but we didn't know the etiology. Now we know the etiology and we can treat it," says Maurer.
"I actually see this as a good model for how we go forward in the world of HFpEF," Maurer says, who also asks whether now we can find other end-phenotypes that are driven by a specific biology that is targetable. Tafamidis has orphan drug status in both the European Union and the U.S., and a high price tag to match. In the U.S., the wholesale price (as of September 2019) is $225,000 per year, or $616 per day.
In a cost-effective analysis that used inputs from the ATTR-ACT trial, investigators found that tafamidis could be expected to add 1.29 quality-adjusted life-years at an incremental cost effectiveness ratio of $880,000 per quality-adjusted life-year gained.12
Given the usual $100,000 threshold for cost-effectiveness used in the U.S., a 92.6% price reduction from $225,000 to $16,563 would be necessary to make tafamidis cost effective. Competition when more agents enter this class of drugs may lead to such a price reduction and those trials are ongoing.
Still, the out-of-pocket costs are a concern for many patients, and polypharmacy and newer more expensive drugs can increase this burden. Cardiology will look at the cost of drugs as a continuing barrier to health care later this year.
There are 40 proteins known to form amyloid fibrils, which can deposit in the extracellular space and disrupt tissue architecture and cause organ dysfunction.1 The vast majority of cardiac amyloidosis is caused by just two of them: light-chain amyloid (AL) and transthyretin amyloid (ATTR).
The nomenclature is simple: The A is for amyloid, followed by the letter or letters referring to the main protein being deposited. For example, light-chain amyloidosis is AL (A for amyloid and L for light chain). Transthyretin amyloidosis is ATTR (A for amyloid and TTR for transthyretin).
Older terms such as primary or secondary amyloidosis, senile amyloidosis and familial amyloid cardiomyopathy are outdated and confusing and should be avoided, according to Ronald Witteles, MD, FACC.
AL amyloidosis is a rare disease with a rapidly progressive clinical course. AL is the most commonly diagnosed form of systemic amyloidosis but is far less commonly found compared to ATTR. The disease arises from overproduction and misfolding of monoclonal immunoglobulin light chains and, left untreated, has a median survival of less than six months when cardiac involvement is prominent.
"AL amyloidosis is not common and is not likely something a community cardiologist will be treating. But it's important to have some understanding of it in order to rule it out on the way to diagnosing ATTR amyloidosis," says Witteles.
The form of cardiac amyloidosis currently getting the most attention is transthyretin amyloid cardiomyopathy (ATTR-CM), an infiltrative heart muscle disease caused by extracellular deposition of insoluble transthyretin amyloid fibrils.
Transthyretin, formerly called prealbumin, is a liver-synthesized plasma protein normally involved in transporting vitamin A (retinol) and the hormone thyroxine throughout the body. TTR exists mainly in a tetrameric state with four noncovalently associated subunits with one protein fitting together with three others to form its normal structure. "It looks like a four-leaf clover," according to Mathew Maurer, MD, FACC.
"Normally, in between the leaves is where the thyroid hormone and vitamin A fit and get carried around and distributed in the bloodstream to tissues," explains Maurer. With ATTR-CM, amyloid fibrils mainly deposit in the interstitial space of the myocardium, leading to increased wall thickness, diastolic dysfunction and arrhythmias.
There are two subtypes of ATTR-CM. Hereditary or variant ATTR is caused by TTR mutations (of which there are about 120), while wild-type ATTR amyloidosis, the most common form of cardiac amyloidosis and previously referred to as senile systemic amyloidosis, is a result of age-related changes in wild-type TTR stability.
"If you're born with a genetic TTR variant, the protein becomes unstable, doesn't fold correctly and form exactly right. Instead of looking like a four-leaf clover, it falls apart and the pieces are floating around in the bloodstream. Those pieces line up and form amyloid and the amyloid gets stuck in the heart," explains Maurer.
Reference
- Benson MD, Buxbaum JN, Eisenberg DS, et al. Amyloid 2020;27:217-22.
References
- Liu C, Witschey WRT, Santangeli P, Han Y. Spontaneous intramyocardial haemorrhage in a patient with wild-type transthyretin cardiac amyloidosis. Eur Heart J 2020;2020: Dec 21:[Epub ahead of print].
- Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: Evolving diagnosis and management: A scientific statement from the American Heart Association. Circulation 2020;142:e7-e22.
- González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015;36:2585-94.
- Castaño A, Narotsky DL, Hamid N, et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J 2017;38:2879-87.
- Shah KB, Mankad AK, Castano A, et al. Transthyretin cardiac amyloidosis in Black Americans. Circ Heart Fail 2016;9:e002558.
- Jacobson DR, Pastore RD, Yaghoubian R, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med 1997;336:466-73.
- Arora S, Patil NS, Strassle PD, et al. Amyloidosis and 30-day outcomes among patients with heart failure. JACC: CardioOncology 2020;2:710-8.
- Joynt Maddox KE, Zhang KW. Cardiac amyloidosis: More than a needle in a haystack. JACC: CardioOnc 2020;2:719-20.
- Witteles RM, Bokhari S, Damy T, et al. Screening for Transthyretin amyloid cardiomyopathy in everyday practice. JACC Heart Fail 2019;7:709-16.
- Geller HI, Singh A, Alexander KM, et al. Association between ruptured distal biceps tendon and wild-type transthyretin cardiac amyloidosis. JAMA 2017;318:962-63.
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018;379:1007-16.
- Kazi DS, Bellows BK, Baron SJ, et al. Cost-effectiveness of tafamidis therapy for transthyretin amyloid cardiomyopathy. Circulation 2020;141:1214-24.
Clinical Topics: Anticoagulation Management, Arrhythmias and Clinical EP, Cardiac Surgery, Cardiovascular Care Team, Dyslipidemia, Heart Failure and Cardiomyopathies, Invasive Cardiovascular Angiography and Intervention, Noninvasive Imaging, Prevention, Valvular Heart Disease, Atherosclerotic Disease (CAD/PAD), Anticoagulation Management and Atrial Fibrillation, Implantable Devices, SCD/Ventricular Arrhythmias, Atrial Fibrillation/Supraventricular Arrhythmias, Aortic Surgery, Cardiac Surgery and Arrhythmias, Cardiac Surgery and Heart Failure, Cardiac Surgery and VHD, Acute Heart Failure, Interventions and Coronary Artery Disease, Interventions and Imaging, Interventions and Structural Heart Disease, Computed Tomography, Echocardiography/Ultrasound, Nuclear Imaging, Hypertension
Keywords: ACC Publications, Cardiology Magazine, Adipose Tissue, African Americans, American Heart Association, Algorithms, Amyloid, Amyloidosis, Anticoagulants, Aortic Valve Stenosis, Arrhythmias, Cardiac, Atrial Fibrillation, Atrial Fibrillation, Biopsy, Blood Proteins, Cardiomyopathies, Carpal Tunnel Syndrome, Contrast Media, Coronary Artery Disease, Diagnostic Errors, Diphosphates, Diphosphonates, Echocardiography, Electrophoresis, Equidae, Exercise Tolerance, Family Characteristics, Feasibility Studies, Gadolinium, Genetic Testing, Genotype, Heart Failure, Heart Ventricles, Hematologic Tests, Hemorrhage, Hospital Mortality, Hospitalization, Hospitals, Hyperlipidemias, Hypertension, Hypertrophy, Immunotherapy, Inpatients, Kidney Diseases, Laboratories, Liver, Magnetic Resonance Spectroscopy, Mutation, Myocardium, Neoplasms, Patient Readmission, Plaque, Amyloid, Pharmaceutical Preparations, Polyneuropathies, Prealbumin, Prevalence, Quality of Life, Radionuclide Imaging, Random Allocation, Risk, Severity of Illness Index, Standard of Care, Stroke Volume, Tachycardia, Supraventricular, Technetium, Thyroxine, Tomography, Emission-Computed, Single-Photon, Transcatheter Aortic Valve Replacement, United States Food and Drug Administration, Universities, Vulnerable Populations
< Back to Listings
