Cover Story | Emerging From the Shadows: Lp(a)

After several decades hovering in the shadows of clinical lipidology, lipoprotein(a) (Lp[a]) is stepping into the spotlight. This move to star status is fueled by accumulating evidence of its contribution as an independent causal risk factor for cardiovascular disease and aortic stenosis, but more so by the promise of new therapeutics that might markedly modify this risk.
Cardiology checked in with two experts in the field to review the evidence and better understand the role the larger cardiology and primary practice communities need to play to understand the role of Lp(a) in atherosclerotic cardiovascular disease (ASCVD) and what to do about it, both now and the near future.
Lipoprotein(a) Epidemiology
Lp(a) was first identified by Norwegian geneticist Kare Berg in 1963.1 Berg recognized that Lp(a) concentration was genetically controlled and, by 1974, had linked the presence of Lp(a) to coronary heart disease.2 Over the years, as assays for Lp(a) improved, additional evidence emerged linking Lp(a) to a range of atherothrombotic and nonatherosclerotic conditions.
Lp(a) is a cholesterol-carrying lipoprotein, made mostly in the liver and consists of an LDL-like lipoprotein and two proteins known as ApoB and Apo(a). Like other apo B-containing lipoproteins, Lp(a) can become trapped within the artery wall where it can participate in the initiation and progression of atherosclerotic plaque. Pathogenic mechanisms of Lp(a) are illustrated in Figure 1.
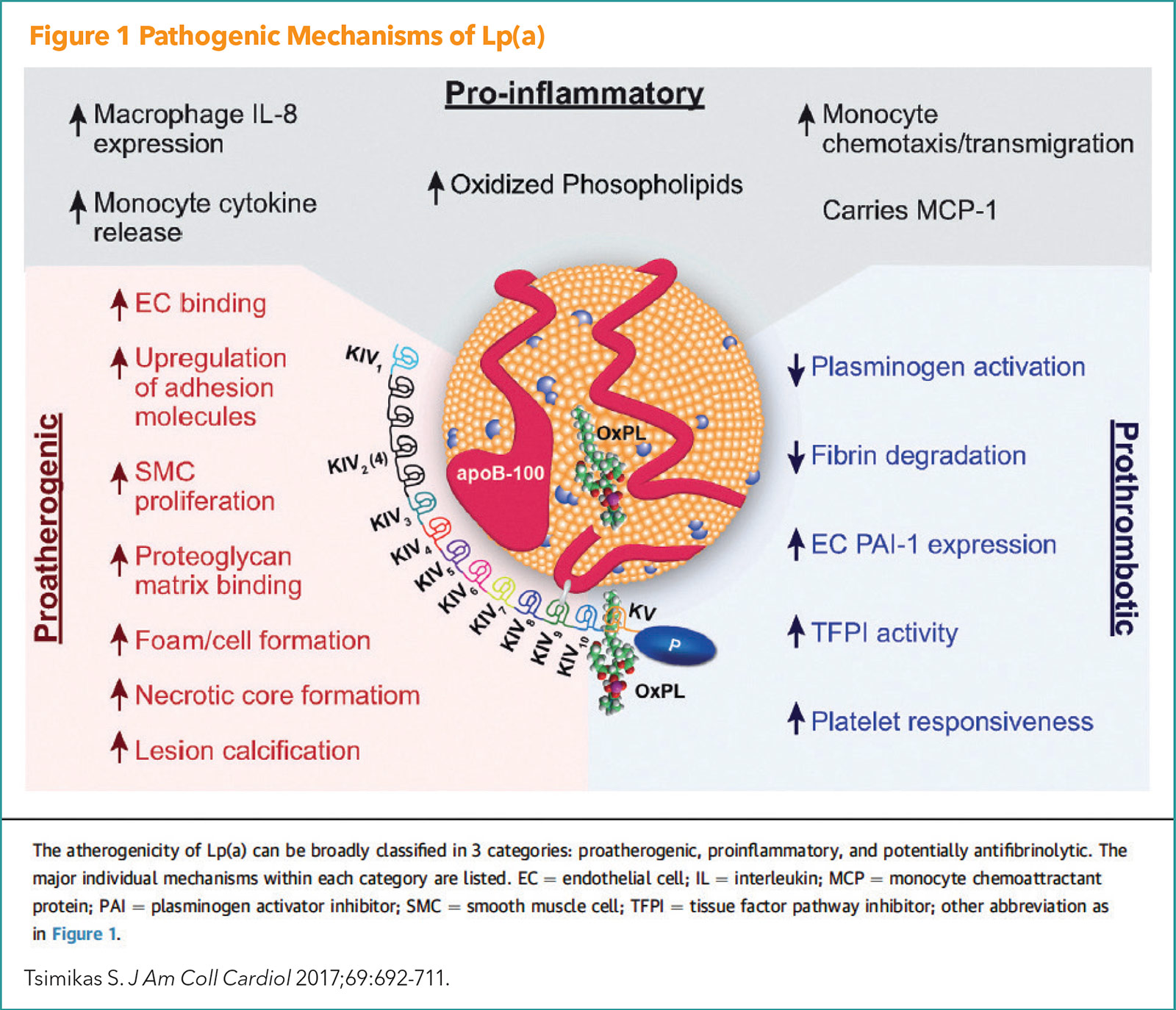
There is consistent evidence from Mendelian randomization and epidemiological studies supporting a causal role for elevated Lp(a) in the development of ASCVD and nonatherosclerotic disease, including calcific aortic valve stenosis and atrial fibrillation.
"The evidence suggests that persons with extreme elevations of Lp(a) – above 150 mg/dL – have a lifetime risk equivalent to persons with heterozygous familial hypercholesterolemia (FH), but this level of Lp(a) is about two- to three-times more prevalent than FH," says Brian A. Ference, MD, MPhil, MSc, FACC.
"Elevated LP(a) is probably the most commonly inherited genetic risk for ASCVD, which is where things get exciting when we start looking at drugs that potently lower it," he adds.
Ference is a professor and director of Research in Translational Therapeutics, and executive director of the Centre for Naturally Randomized Trials at the University of Cambridge, UK.

What Gets Measured Gets Managed
When should clinicians measure Lp(a)? A lot more often than they do now, according to the experts.
"If you go into the coronary care unit and see someone who is 40 years old with an acute myocardial infarction (MI), you need to know the level of their Lp(a), because those are the people who have Lp(a) levels of 200 and 300 mg/dL or even higher," says Steven E. Nissen, MD, MACC.
Nissen is the chief academic officer at the Sydell and Arnold Miller Family Heart, Vascular and Thoracic Institute, and holds the Lewis and Patricia Dickey Chair in Cardiovascular Medicine at the Cleveland Clinic.
There are three compelling reasons to obtain at least a one-time measurement of Lp(a) in most patients, suggests Ference, even though at present there is no effective means to lower it.
Of note, all three reasons are integrated into a new guideline for managing Lp(a) from the European Society of Atherosclerosis and scheduled to be released at its meeting in late May. Ference is an author of the guideline.
"One, Lp(a) needs to be measured so it can be included in new algorithms to estimate exactly how much Lp(a) increases risk, and by extension, the degree of benefit from lowering it when a therapy becomes available," says Ference.
Two, after the level of increased risk from elevated Lp(a) is better understood, it is possible to minimize the Lp(a)-related risk, despite no real decrease in the Lp(a) level, by intensifying risk factor modification. This will be addressed in the new EAS guideline with an algorithm that will help clinicians understand how much the risk is increased and how much to intensify treatment, for example with additional lowering of LDL-C or blood pressure. Currently, there is little guidance, and "we need more specific and actionable information" says Ference.
Three, measuring Lp(a) will help increase awareness among family members. By most estimates, elevated Lp(a) is inherited in 70% to 90% of cases. Considering the high likelihood that a child or other family member also could have high levels of Lp(a), this awareness and testing can lead to earlier treatment and intensification of lowering LDL-C and blood pressure to prevent ASCVD. And potentially lead to treatment with new therapies when they become available.
Cumulative lifetime exposure to higher levels of LDL-C determines cardiovascular risk, according to Mendelian randomization studies. While this is also believed to be true for Lp(a), this has not been proven yet.
If the trials of Lp(a) show the benefit of lowering it starting later in life is smaller than the benefit of lifelong lowering of LDL-C, "we can conclude the effect of elevated Lp(a) levels increases over time just like for LDL-C," explains Ference. Because Lp(a) is largely inherited, this could lead to a recommendation to measure it early in life, perhaps in the teens, to identify those at "very high risk." And provide an earlier start to reducing risk by getting LDL-C levels really low, controlling other risk factors very well, and "hopefully one day soon, treat their Lp(a)," Ference says.

Brian A. Ference, MD, MPhil, MSc, FACC
This hints at an especially exciting future for the prevention of ASCVD – with a combination of drugs with the potential to lower LDL-C and Lp(a). For example, a PCSK9 inhibitor like inclisiran that lowers LDL-C with an injection twice a year and a drug that lowers Lp(a) potently in patients for whom it is indicated. Ference notes that the potential for this approach to "almost eliminate atherosclerosis from society with early treatment is pretty exciting."
"It is absolutely crucial that patients have their Lp(a) level measured, particularly those with premature cardiovascular disease or strong family histories," states Nissen. In fact, "almost everybody should now have Lp(a) measured, probably in their twenties, to know if they're at risk for ASCVD and can get proper guidance."
How High is Too High? Next Steps?
Lp(a) has a skewed distribution in most populations, with a long tail of individuals with higher concentrations of Lp(a). Most individuals have normal levels and the mean and median concentrations in the general population are well below the levels associated with increased risk. This asymmetric distribution means Lp(a) will be a very important risk factor only for the approximately 20% of those in the tail of the distribution. Unlike with blood pressure and weight, for example, which are normally distributed in the population such that the median level is quite high and everyone is likely to benefit from a reduction.
This provides a hint as to why drugs that lower Lp(a) moderately might have failed to reduce risk in clinical trials. Similar with what is seen with LDL-C, it is the absolute decrease in Lp(a) level that reduces risk, not the percentage its reduced. And because the median level of Lp(a) in the population is low, even lowering it by 50% does not provide a very big absolute reduction or benefit.
Therefore, the protocol for the ongoing HORIZONS trial testing a new drug to lower Lp(a) specifies enrolling patients above the 90th percentile of Lp(a) levels. The rationale is to have the ability to achieve large absolute reductions in Lp(a) to definitively determine whether this translates into a large, clinically important decrease in risk, says Ference, who is not involved in the trial but has consulted for Novartis.
Furthermore, it is the magnitude of the reduction that determines the benefit. This is because the risk increases continuously with increasing Lp(a) levels. A level of 50 means moderate risk and 150 means a very high-risk equivalent similar with that associated with FH.
But 50 and 150 what? It turns out that's a source of confusion too. (The answer is mg/dL.)
Some labs report Lp(a) as mass or weight in milligrams per deciliter (mg/dL). Other labs report the number of particles, or concentration, in nanomoles per liter (nmol/L). Nissen suspects that ultimately, nmol/L will be the preferred unit of choice for measuring Lp(a).

Steven E. Nissen, MD, MACC
"Clearly, it's not reasonable to look at Lp(a) as a dichotomous risk variable because risk increases with increasing Lp(a) levels regardless of the threshold chosen," says Ference, but also because we don't even have a standard for measuring it.
Nissen calls them "risky levels" rather than risk levels. "Most people believe the threshold where you start to worry about patients is about 50 mg/dL, which is equivalent to about 125 nmol/L," he says. Unfortunately, it is easy to get confused because both units are used in the literature and converting nmol to mg/dL is not easy or precise. "Saying that 50 mg/dL is about 125 nmol/L is a rough conversion, not an exact conversion," notes Nissen.
The distribution of Lp(a) in most populations is similar, but there are some big racial and ethnic differences in Lp(a) distribution. Based on data from the MESA trial, Whites, Chinese and Hispanics/Latinx have mean levels around 13 mg/dL.3 Mean levels in Blacks, however, hover around 35 mg/dL.
"Some people believe the LPA gene originated in Africa and then migrated out of Africa, and somehow persisted at a higher frequency in the original population. But I don't think anybody really knows for sure," notes Nissen.
In his weekly clinic where he manages individuals with elevated Lp(a), Nissen tends to use a cut-off of 60 mg/dL. "Using 60 mg/dL as a cut-off, there are about 64 million people in the U.S. alone with high Lp(a) who might benefit from Lp(a) lowering. Worldwide, the number is 1.4 billion," he notes.4
Targeted Lp(a) Lowering Drugs on the HORIZONS

There are currently two types of gene-silencing Lp(a)-lowering drugs in development – antisense oligonucleotides (ASO) and short interfering RNAs (siRNA).
ASOs lower Lp(a) by inhibiting the production of apolipoprotein(a) in the hepatocyte. They target the hepatic LPA messenger RNA (mRNA) reducing plasma levels of Lp(a).
"The ASO approach to gene silencing involves single-stranded DNA that binds to messenger RNA, which is subsequently degraded so the message to produce apo(a) never gets translated," explains Nissen. "It's as if you're turning off the LPA gene responsible for elevated Lp(a) levels."
Leading the Lp(a)-lowering race is the ASO pelacarsen, which in a phase 2 study reduced Lp(a) by as much as 80%.5 The agent is currently being tested in the phase 3 Lp(a)HORIZONS cardiovascular outcomes trial.
Lp(a)HORIZONS aims to definitively assess the safety and efficacy of pelacarsen in 8,000 patients worldwide. Importantly, eligible participants will have Lp(a) levels ≥70 mg/dL at the screening visit and established coronary artery disease (MI or ischemic stroke ≥3 months but ≤10 years from screening/randomization) or clinically significant symptomatic peripheral artery disease.
Participants will be stratified according to their screening Lp(a) level (≥70 mg/dL and ≥90 mg/dL) and receive optimal background therapy, including statins, and be randomized to four years of therapy with either pelacarsen 80 mg or matching placebo, both given by subcutaneous injection once monthly.
The trial is designed to demonstrate the superiority of pelacarsen compared with placebo in reducing the risk of an expanded major adverse cardiovascular events composite (cardiovascular death, nonfatal MI, nonfatal stroke and urgent coronary revascularization requiring hospitalization) in the overall study population with established ASCVD and Lp(a) ≥70 mg/dL and in the subpopulation with established ASCVD and Lp(a) ≥90 mg/dL.
The minimum follow-up in the double-blind period is required to be 2.5 years; the overall trial duration is expected to be approximately 4.25 years, with a readout not expected until 2025.
"The U.S. Food and Drug Administration has appropriately said that no drug is going to be approved for lowering Lp(a) until somebody can show that lowering Lp(a) reduces morbidity and mortality. That's exactly what we're doing with Lp(a)HORIZONS," says Nissen, the chair of the trial.
In 2017, Novartis entered into an option agreement to develop and commercialize pelacarsen (previously known as AKCEA-APO(a)-LRx), announcing in 2019 that it was exercising its option to license the rights to the drug and providing a $150 million payout to Ionis Pharmaceuticals.
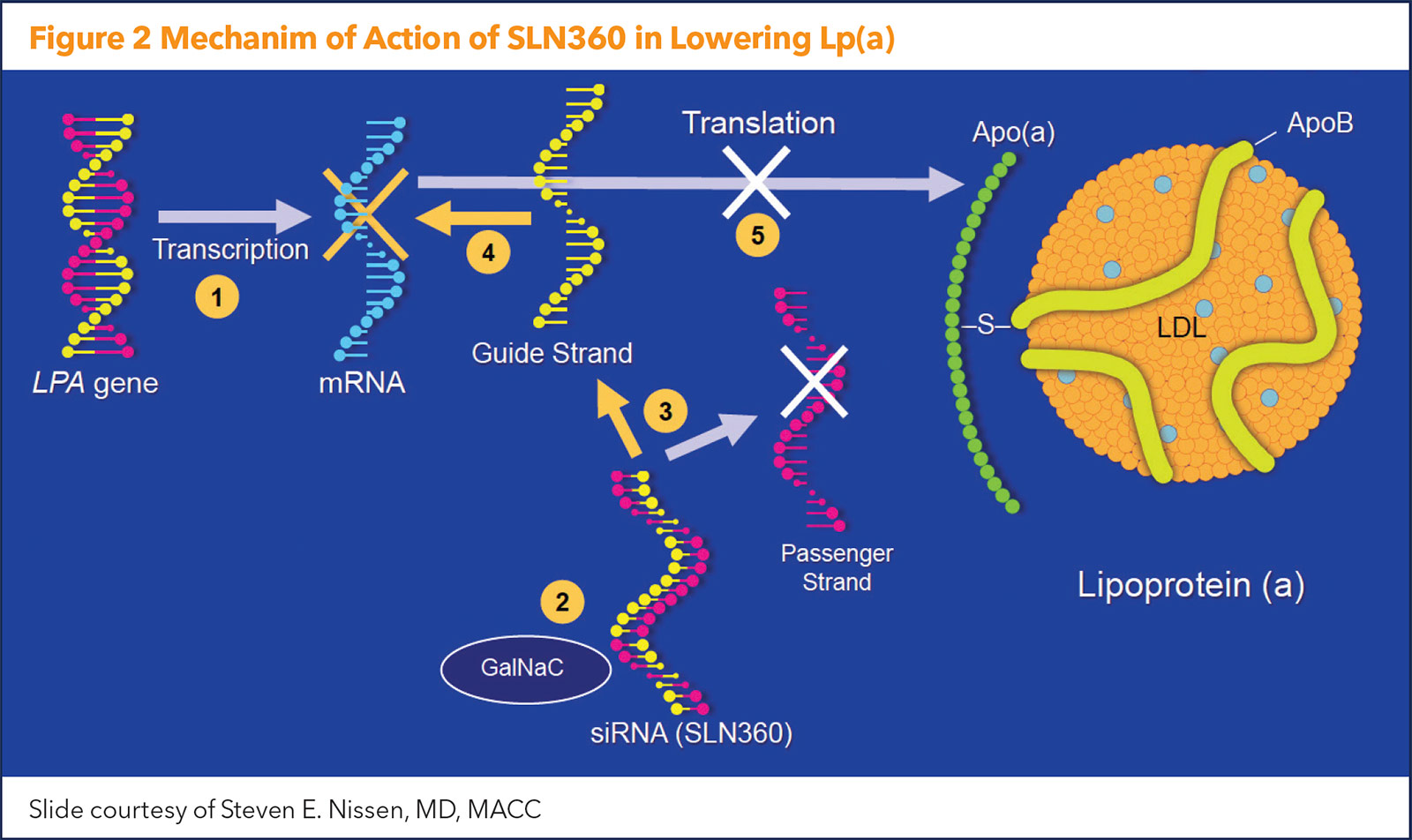
There are currently two SiRNAs in development: SLN360 (Silence Therapeutics, London, UK) and olpasiran (Amgen, Thousand Oaks, CA). Both are designed to directly inhibit LPA messenger RNA translation in hepatocytes and potently reduce plasma Lp(a) concentration. Results from a phase 2 dose-finding study of olpasiran are expected within the year.
The newest data on Lp(a) lowering with an siRNA comes from the APOLLO trial, presented by Nissen as a Late-Breaking Clinical Trial at ACC.22. The study was simultaneously published in JAMA, with an editorial comment provided by Ference.6,7 Figure 2 illustrates the mechanism of action of the siRNA agent.
APOLLO was a phase 1 dose-finding trial that included 32 adults (mean age, 50 years; 53% female) with Lp(a) plasma concentration of ≥150 nmol/L at screening and no known clinically overt cardiovascular disease. All were randomly assigned to placebo or single doses of SLN360 at 30 mg, 100 mg, 300 mg or 600 mg, administered subcutaneously.
The median baseline Lp(a) concentration was 224 nmol/L – clearly well into the "risky" level. After a single dose, the maximal median change in Lp(a) was –20 nmol/L in the placebo arm and –89 nmol/L, –185 nmol/L, –268 nmol/L and –227 nmol/L in the 30-mg, 100-mg, 300-mg and 600-mg SLN360 groups, respectively. This translated to maximal median percent changes of –10%, –46%, –86%, –96% and –98% for the placebo group and ascending dose groups, respectively (Figure 3).
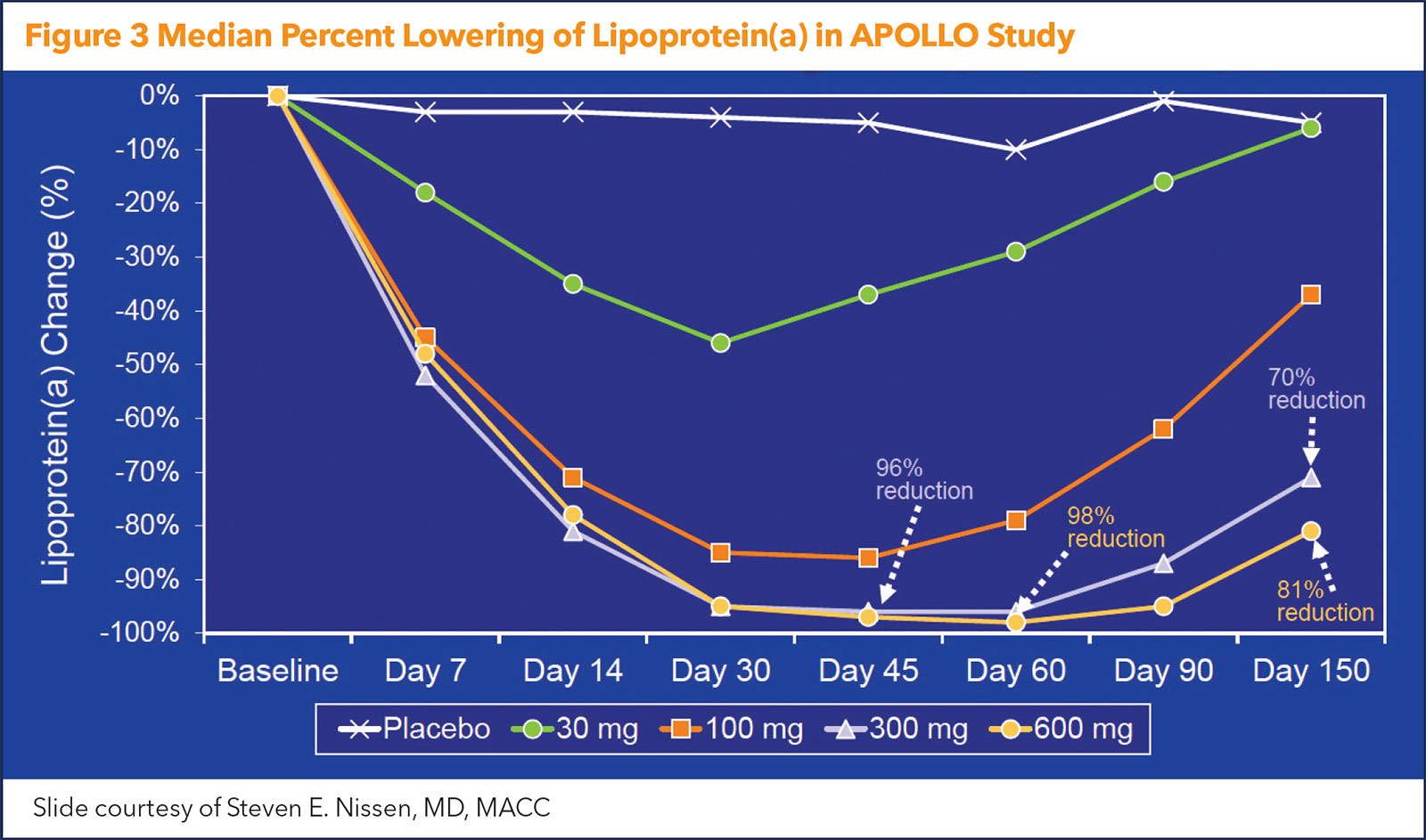
The duration of Lp(a) lowering was dose dependent, but five months after treatment, Lp(a) levels were still 71% to 81% below baseline at the highest doses. As an added plus, in the highest dose groups, LDL-C was reduced 20% to 25%.
"We thought it would work, but we were surprised by the magnitude and the duration of the effect," says Nissen, the study's chair.
For both pelacarsen and SLN360, the most common side effect was temporary soreness at the injection site. "They all appear to cause injection site reactions, not unlike after getting a COVID vaccine. But if you get an injection site reaction and also protection from a deadly disease, most people won't even think about it," says Nissen.
Which therapeutic approach might ultimately prevail is anyone's guess, says Nissen. "When you're taking on a disease we've never been able to treat, you want to have multiple shots on goal, and you hope one of them makes into the back of the net."
All told, it's an exciting time for the field. "We are now at a really pivotal turning point in the treatment of a disease that was previously considered an untreatable abnormality," says Nissen. "Optimism is warranted."
This article was authored by Debra L. Beck, MSc.
References
- Berg K. A new serum type system in man--the Lp system. Acta Pathol Microbiol Scand 1963;59:369-82.
- Reyes-Soffer G, Ginsberg HN, Berglund L, et al. Lipoprotein(a): A genetically determined, causal, and prevalent risk factor for atherosclerotic cardiovascular disease: A scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol 2022;42:e48-e60.
- Guan W, Cao J, Steffen BT, et al. Race is a key variable in assigning lipoprotein(a) cutoff values for coronary heart disease risk assessment: the Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol 2015;35:996-1001.
- Varvel S, McConnell JP, Tsimikas S. Prevalence of elevated lp(a) mass levels and patient thresholds in 532 359 patients in the United States. Arterioscler Thromb Vasc Biol 2016;36:2239-45.
- Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med 2020;382:244-55.
- Nissen SE, Wolski K, Balog C, et al. Single ascending dose study of a short interfering RNA targeting lipoprotein(a) production in individuals with elevated plasma lipoprotein(a) levels. JAMA 2022;April 3:[Epub ahead of print].
- Ference BA. The potential clinical benefit of lowering lipoprotein(a). JAMA 2022;April 3:[Epub ahead of print].
Clinical Topics: Arrhythmias and Clinical EP, Cardiovascular Care Team, Dyslipidemia, Prevention, Valvular Heart Disease, Atrial Fibrillation/Supraventricular Arrhythmias, Advanced Lipid Testing, Lipid Metabolism, Nonstatins, Novel Agents, Primary Hyperlipidemia
Keywords: ACC Publications, Cardiology Magazine, Primary Prevention, Cholesterol, LDL, PCSK9 protein, human, Proprotein Convertase 9, Cardiovascular Diseases, Atrial Fibrillation, Coronary Care Units, Blood Pressure, Plaque, Atherosclerotic, Mendelian Randomization Analysis, Risk Factors, Heart Disease Risk Factors, Lipoprotein(a), International Classification of Diseases, Atherosclerosis, Hyperlipoproteinemia Type II, Epidemiologic Studies, Coronary Disease, Family, Myocardial Infarction, Aortic Valve Stenosis, Algorithms, Arteries, Liver, Apolipoproteins A, Apolipoproteins B, Middle Aged, COVID-19 Vaccines, Injection Site Reaction, RNA, Small Interfering, RNA, Small Interfering, RNA, Messenger, Hepatocytes
< Back to Listings
